Este es un trabajo sumamente importante con implicaciones tanto para la Enfermedad Alzheimer como para el uso de transfusiones de plaquetas y sangre y transplantes de órganos y médula ósea.Los resultados de este trabajo ameritan la atención de los médicos, autoridades de salud y péublico en general.
Resumen
El riesgo de enfermedad iatrogénica a menudo se subestima como una preocupación en los procedimientos médicos contemporáneos, que abarcan el trasplante de tejidos y órganos, las terapias con células madre, las transfusiones de sangre y la administración de productos derivados de la sangre. En este contexto, a pesar de la creencia predominante de que la enfermedad de Alzheimer (EA) se manifiesta principalmente en formas familiares y esporádicas, la investigación de Singh,et.al.(Stem Cell Reports.Published:March 28, 2024DOI:https://doi.org/10.1016/j.stemcr.2024.02.012) revela, una variante trasplantable inesperada de la EA en un contexto preclínico, que potencialmente indica una transmisión iatrogénica en pacientes con EA. Mediante el trasplante adoptivo de células madre de médula ósea de un donante que transportan un transgén de proteína precursora de amiloide humana (APP) mutante en animales receptores normales o deficientes en APP, observaron un rápido desarrollo de características patológicas de la EA. Estas características patológicas se aceleraron significativamente y surgieron entre 6 y 9 meses después del trasplante e incluyeron integridad comprometida de la barrera hematoencefálica, neoangiogénesis vascular cerebral elevada, niveles elevados de β-amiloide asociado al cerebro y deterioro cognitivo. Además, sus hallazgos subrayan la contribución de la carga de β-amiloide que se origina fuera del sistema nervioso central a la patogénesis de la EA dentro del cerebro. Singh,y colegas concluyen,correctamente, que el trasplante de células madre de donantes que albergan un alelo mutante patógeno puede transferir eficazmente enfermedades del sistema nervioso central a receptores sanos, reflejando la patogénesis observada en el donante. En consecuencia, estas observaciones abogan por la secuenciación genómica de muestras de donantes antes de las terapias de trasplante de tejidos, órganos o células madre, así como de las transfusiones de sangre y la administración de productos derivados de la sangre, para mitigar el riesgo de enfermedades iatrogénicas.
=> Recibir por Whatsapp las noticias destacadas
En Detalle
La utilización de productos derivados de la sangre, transfusiones de sangre, transfusiones de plaquetas, trasplantes de órganos y trasplantes de médula ósea, así como terápias emergentes personalizadas basadas en células y tejidos, como la transferencia adoptiva de células madre pluripotentes inducidas (iPS), representan intervenciones clínicas críticas. No obstante, existen preocupaciones inminentes con respecto a su seguridad. Recientemente, se ha propuesto que la enfermedad de Alzheimer (EA) es una enfermedad similar a los priones en la que la patología puede transmitirse de forma similar a los priones; por ejemplo, la transferencia de una proteína mal plegada es suficiente para provocar una enfermedad en el receptor. La idea de que los péptidos β-amiloide (Aβ) mal plegados son partículas infecciosas proteicas se informó anteriormente cuando extractos de cerebro de pacientes con EA fueron capaces de inducir amiloidosis cerebral cuando se transfirieron a modelos de ratón y primates . Los informes sobre la transferencia de patología de la EA en humanos se han asociado con el uso de hormona de crecimiento derivada de la hipófisis de cadáveres humanos (c-hGH) contaminada. Los pacientes que habían recibido c-hGH contaminada murieron de enfermedad de Creutzfeldt-Jakob (ECJ) iatrogénica, pero, tras la investigación, se informó que estos pacientes también tenían Aβ inmunorreactivo sustancial y un mayor número de microvasos en el cerebro,
que es una patología asociada a la EA, no a la ECJ. Posteriormente, se encontró que algunos lotes de c-hGH, a los que fueron expuestos pacientes con ECJ con patología Aβ, tenían niveles sustanciales de proteínas Aβ40, Aβ42 y tau . Sin embargo, en el caso de estos estudios de siembra de Aβ, se supone que el Aβ endógeno del receptor contribuye a la enfermedad en un proceso en cascada mal plegado. De todos modos, la capacidad de transmisión de enfermedades genera preocupación para muchas de las terapias basadas en células, incluido el trasplante de médula ósea o el tratamiento con sangre del cordón umbilical o células madre de embriones humanos. Actualmente, muchos países aprueban el uso de médula ósea y células madre como terapias contra el cáncer , y hay mucha investigación sobre su uso para los trastornos del sistema nervioso central (SNC), incluida la EA , la enfermedad de Parkinson , la esclerosis múltiple y la lesión de la médula espinal .
La comprensión tradicional de la EA se centra en la acumulación de Aβ en el cerebro. Anteriormente, se pensaba que este Aβ se originaba principalmente en el propio cerebro . Sin embargo, investigaciones recientes sugieren que las especies de Aβ en el torrente sanguíneo pueden cruzar la barrera hematoencefálica (BHE), contribuyendo significativamente a la patología de la EA . Estos hallazgos, basados predominantemente en modelos con un gen de la proteína precursora de amiloide (APP) intacto en los receptores, no determinan de manera concluyente si el Aβ generado dentro del SNC es paralelo al proceso observado en las enfermedades priónicas, ni exploran completamente vías alternativas, como las que involucran inflamación y mediadores inflamatorios, que podrían influir en la acumulación de Aβ endógeno en el SNC.
Los datos anteriores del lasoratorio de Singh y colegas demuestran que Aβ proporciona una señal proangiogénica para la vasculatura cerebral y que Aβ puede ingresar posteriormente al cerebro por difusión después de que la BHE se interrumpe cuando las uniones estrechas desaparecen durante la división celular . La principal fuente potencial de APP periférica que contribuye a este mecanismo son las plaquetas que se originan a partir de megacariocitos formados a partir de células madre hematopoyéticas (HSC) . Tras la activación plaquetaria, la actividad de la enzima β-secretasa aumenta en sus membranas, lo que resulta en la producción de especies de Aβ solubles . Aunque las plaquetas que se originan en las HSC son la principal fuente de Aβ periférico, otras fuentes como los fibroblastos de la piel, los músculos esqueléticos y las células del músculo liso cerebrovascular también pueden producir Aβ .
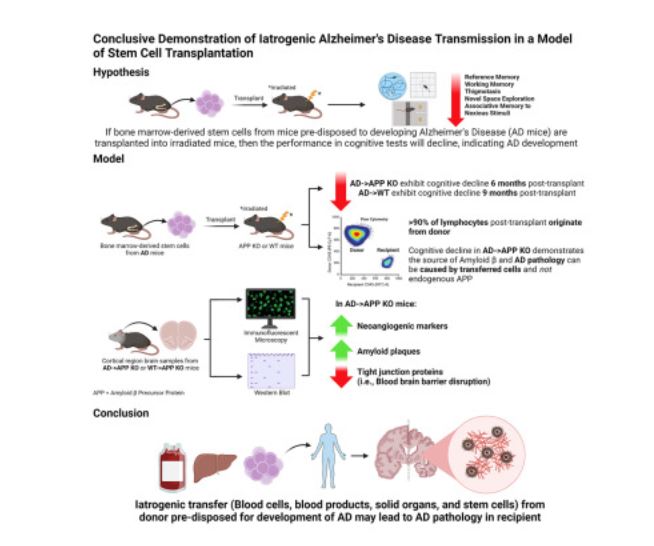
Hasta la fecha, ninguno de los estudios anteriores se ha esforzado por cumplir los postulados de Koch para establecer la etiología microbiológica de la infección y la enfermedad. En este estudio, Singh y colegas investigaron si las características patológicas de la EA pueden transferirse mediante la repoblación de los compartimentos HSC de un modelo de ratón knockout de tipo salvaje o deficiente en APP con células de médula ósea (BMC) de donantes que albergan el gen APP humano mutante sueco (APPKM67/671NL) vinculado a la EA familiar de inicio temprano.
Aparte de las enfermedades priónicas o las encefalopatías espongiformes transmisibles (EET), este parece ser el primer informe definitivo de que el trasplante celular puede transferir de forma adoptiva una enfermedad del SNC, y esta ocurrencia es independiente del Aβ endógeno. Además, para imitar el entorno clínico, también examinaron si la patología de la EA se puede establecer en animales APP de tipo salvaje (WT) después de la transferencia adoptiva de BMC que contienen el gen APP mutado que causa la enfermedad y descubrieron que estas observaciones tienen una correlación directa a escenarios clínicos donde la enfermedad iatrogénica puede ser un riesgo.
Este estudio proporciona nuevos conocimientos sobre el papel de la APP periférica en el desarrollo de la EA, desafiando nuestra comprensión actual. Además, subraya las preocupantes implicaciones para la transferencia iatrogénica de otras enfermedades.
Este estudio cuestiona la comprensión predominante de que la EA es exclusivamente familiar o esporádica. Utilizando un modelo preclínico, Singh y colegas demostraron una forma trasplantable de EA, lo que sugiere una posible transmisión iatrogénica en pacientes con EA. Los investigadores examinaron específicamente el potencial de los trasplantes de células madre de médula ósea en la transmisión de la EA en un modelo familiar de esta enfermedad mortal. Descubrieron que la transferencia de médula ósea de ratones AD a ratones APP KO provocaba un deterioro cognitivo, así como depósitos extracelulares de Aβ y disfunción de la BHE. En particular, encontraron que los ratones quiméricos AD→APP KO acumulan Aβ humano en el cerebro procedente exclusivamente del donante, muestran una mayor expresión del marcador neoangiogénico CD105 y muestran una expresión disminuida de los TJP ocludina y ZO1. Los datos demuestran que el trasplante de médula ósea, que contiene HSC, de un ratón donante que sobreproduce una APP humana mutante, puede transferir la patología de la EA en un animal receptor que no sintetiza APP endógena. Además, la tasa de formación de la enfermedad en el modelo trasplantable es mucho más rápida que en los ratones AD, ya que se establece en los receptores de médula ósea AD en 6 meses en comparación con los 12 meses en los ratones transgénicos AD .
De hecho, existe evidencia de que los receptores de trasplantes, incluidos los de órganos sólidos y CMH, tienen una mayor incidencia de diversas enfermedades neurológicas. Para los receptores de trasplantes de órganos sólidos, las complicaciones del SNC son relativamente comunes e incluyen déficits neurológicos tanto focales como difusos. Estas complicaciones pueden deberse a diversas causas, incluidas infecciones, toxicidad de medicamentos, eventos cerebrovasculares, trastornos metabólicos y cáncer. En particular, se informa que la incidencia de complicaciones neurológicas en los receptores de trasplantes de riñón es de alrededor del 10% al 21%, que es mayor que en la población general. Los factores que contribuyen a estas complicaciones incluyen la necesidad de múltiples fármacos, disminución de la inmunidad celular, enfermedad aterosclerótica acelerada y anomalías metabólicas frecuentes .
En el contexto específico del trasplante de células madre hematopoiéticas (TCMH) para la leucemia linfoblástica aguda pediátrica, las complicaciones neurológicas, tanto agudas como a largo plazo, también son comunes y contribuyen a una morbilidad y mortalidad significativas. La incidencia de neurotoxicidad en niños después de un TCMH oscila entre el 11% y el 59%. Un estudio post mortem de 180 receptores de TCMH, incluidos adultos y niños, encontró que el 90% tenía evidencia de anomalía del SNC y, en el 17% de los casos, esta fue la causa de la muerte. Además, los resultados del TCMH son peores para los pacientes que desarrollan neurotoxicidad aguda. La mayoría de los estudios sobre los efectos neurológicos del TCMH en niños se han centrado en la neurotoxicidad aguda, pero, a medida que más niños sometidos a TCMH por leucemia linfoblástica aguda se convierten en sobrevivientes a largo plazo, es cada vez más importante comprender las consecuencias neurológicas a largo plazo de esta modalidad de tratamiento. La radioterapia y la quimioterapia, como parte del tratamiento del TCMH, pueden agravar la lesión neurológica y tener efectos profundos en la maduración cerebral y la función cognitiva .
Los ratones APP KO no producen Aβ endógeno en ningún tejido u órgano. Si las teorías actuales relacionadas con la causa de la EA fueran correctas, por ejemplo, que el Aβ derivado del cerebro causa la patología de la EA, entonces la reconstitución de los ratones APP KO con HSC de la EA no debería haber inducido la patología de la EA. Los hallazgos de este estudio establecen que el Aβ derivado de HSC puede iniciar una patología vascular que conduce a la alteración de la BHE y eventualmente a la patología de la EA, y los hallazgos de este estudio establecen que el Aβ derivado periféricamente puede iniciar la patología vascular y la alteración de la BHE.
La versión de mutación sueca de la expresión del transgén precursor de amiloide humano está impulsada por el promotor priónico que impulsa la expresión en una variedad de tejidos, incluidas las células sanguíneas, y suponemos que esta es la fuente de la APP que se origina en la célula HPC del donante.
Una cantidad significativa del Aβ periférico se encuentra en las plaquetas circulantes que se originan a partir de megacariocitos. En los ratones AD (Tg2576), se sabe que la médula ósea y las HSC que portan la APP mutante humana producen en exceso Aβ en los megacariocitos . Se sabe que el Aβ generado a partir de plaquetas activadas existe en forma soluble y esta propiedad puede permitir que sean transportados desde otro lugar del cuerpo al SNC a través de los vasos cerebrales. Otra forma de que el Aβ soluble entre al SNC es a través de las plaquetas que circulan en los vasos cerebrales .
Las uniones estrechas que se encuentran en la capa endotelial de los vasos cerebrales tienen en sus proximidades grandes cantidades de colágeno, un activador plaquetario, especialmente en la membrana basal de los vasos cerebrales . Este colágeno puede provocar la activación de las plaquetas y la liberación de Aβ en los vasos cerebrales. El Aβ liberado activa la división celular y la vasculogénesis, lo que conduce a la degradación de la integridad de la BHE, lo que permite que el Aβ cruce libremente la BHE y entre al SNC por difusión, donde se deposita en placas amiloides . El receptor de productos de glicación avanzada (RAGE) es un receptor multiligando que también regula la entrada de Aβ periférico al cerebro. Se une a Aβ soluble en el rango nanomolar, y su expresión está regulada positivamente por la presencia de Aβ y también está implicado en la descomposición de la BHE y la patogénesis de la EA . Otra posibilidad de transferencia de Aβ al SNC podría ser como resultado de la alteración fisiológica de la BHE relacionada con la edad. Esta descomposición podría permitir la transferencia del Aβ soluble al cerebro . Una vez que se inicia la patología de la EA, puede desencadenar una mayor vasculogénesis y degradación de la BHE. Sin embargo, varios otros tipos de células sanguíneas también expresan APP en el ratón Tg2576; por lo tanto, se necesitan estudios futuros para abordar esto.
Uno de los posibles resultados de este estudio es estimular el campo para alejarse del dogma central convencional de la patología de la EA, que afirma que la acumulación de Aβ derivado del cerebro, específicamente producido por las neuronas, es la causa de la enfermedad. Este estudio demuestra la contribución del Aβ, generado fuera del cerebro, en el establecimiento de la enfermedad. Esto puede brindar una oportunidad para el desarrollo de nuevos biomarcadores para la EA. La producción de patología de EA en ratones AD→APP KO también respalda el concepto de que la EA puede transferirse de un individuo a otro, lo que da como resultado el descubrimiento de una forma trasplantable de AD que es distinta de las formas esporádicas o familiares de la enfermedad. Esto sugiere la importancia de la detección de biomarcadores y la secuenciación de los genomas de donantes de células, tejidos y órganos candidatos para alelos nocivos asociados a enfermedades para eliminar la posibilidad de transferencia de enfermedades antes de utilizarlos como donantes para cualquier terapia de trasplante, incluido el trasplante de tejidos y órganos. así como trasplantes de médula ósea, transfusiones de plaquetas y sangre o terapias basadas en células madre.
Sin embargo, es importante reconocer las limitaciones de este trabajo. Si bien los resultados del estudio que utilizó el modelo Tg2576 AD y ratones APP KO brindan información valiosa, se debe tener precaución al trasladar estos hallazgos a los receptores de trasplantes de células madre humanas. El modelo Tg2576, con expresión de APP mutante bajo el control del promotor de la proteína priónica de hámster, genera un alto nivel de patología que no se observa típicamente en pacientes humanos con EA familiar. El uso de un promotor heterólogo y receptores completamente deficientes en APP, una condición rara en humanos, agrega capas metodológicas adicionales que pueden no reflejar directamente las condiciones en humanos, aunque establece, por primera vez, que es necesaria APP mutante derivada exclusivamente de donantes. y suficiente para causar enfermedad en los receptores.
Dadas estas diferencias, es posible que los hallazgos del estudio no se correspondan directamente con la experiencia clínica del trasplante de células madre y sus posibles implicaciones neurológicas. Es posible que la adopción de un modelo murino no capture completamente la complejidad de la EA en humanos, y se deben reconocer las variaciones en la respuesta inmune y los factores genéticos entre especies. Además, las características patológicas observadas, aunque se parecen a la EA, pueden no replicar con precisión la naturaleza multifacética de la enfermedad en humanos. En particular, la forma de EA estudiada aquí es familiar, lo que puede limitar la transferibilidad de los resultados a los humanos, ya que las formas familiares son más raras que las esporádicas. Sin embargo, la observación adicional en este estudio que demuestra la transferencia del fenotipo AD de donantes de médula ósea modelo Tg2576 AD a receptores WT sanos reduce la posibilidad de que nuestros datos no encuentren sus secuelas humanas.
También es importante reconocer el contexto más amplio de las complicaciones neurológicas relacionadas con los trasplantes. Como lo demuestra la mayor incidencia de enfermedades neurológicas en los receptores de trasplantes, tanto en entornos de trasplante de órganos sólidos como de células madre hematopoiéticas, existe una clara indicación de que los procedimientos de trasplante pueden provocar importantes complicaciones del SNC. Estos incluyen una amplia gama de problemas derivados de infecciones, toxicidades de medicamentos, anomalías metabólicas, eventos vasculares e inmunológicos y la propia enfermedad primaria. Por lo tanto, si bien los hallazgos específicos del estudio podrían no traducirse directamente en casos humanos, sí contribuyen a un creciente conjunto de conocimientos sobre los riesgos neurológicos potenciales asociados con varios tipos de trasplantes. Esta información es crucial para el desarrollo de estrategias para mitigar estos riesgos y mejorar los resultados de los pacientes tanto en poblaciones pediátricas como adultas.
En conclusión, aquí se ha comenzado a abordar la subestimación del riesgo de enfermedades iatrogénicas en las prácticas médicas contemporáneas, incluidos los trasplantes de tejidos y órganos, las terapias con células madre, las transfusiones de sangre y la administración de productos derivados de la sangre. Contrariamente a las creencias predominantes sobre la EA que ocurre únicamente en formas familiares o esporádicas, este estudio revela una forma inesperada trasplantable de EA en un modelo preclínico, lo que sugiere una posible transmisión iatrogénica en pacientes con EA. El trasplante adoptivo de BMC de donantes que albergan un transgén APP humano mutante en animales receptores WT sanos y con deficiencia de APP dió como resultado el rápido desarrollo de características patológicas de la EA. Estos incluyeron integridad comprometida de la BHE, aumento de la neoangiogénesis vascular cerebral, niveles elevados de Aβ asociados al cerebro y deterioro cognitivo que se acelera y ocurre dentro de los 6 a 9 meses posteriores al trasplante. Además, los hallazgos sugieren que la acumulación de Aβ que se origina externamente al SNC contribuye a la patología de la EA.
Si bien el estudio proporciona información valiosa, es esencial realizar más investigaciones con sujetos humanos para validar la aplicabilidad de estos hallazgos en entornos clínicos. Sin embargo, dados los hallazgos de este estudio, la recomendación propuesta para la detección genómica de muestras de donantes antes de las terapias de trasplante o la transferencia de productos derivados de la sangre requiere una deliberación cuidadosa.