Ronald Palacios Castrillo, M.D.,PhD
=> Recibir por Whatsapp las noticias destacadas
Los ratones del laboratorio de Aaron Burberry en la Case Western Reserve University seguían muriendo. Genéticamente, eran idénticos a los ratones de otras instituciones del país. Misma cepa, misma mutación, mismos resultados esperados. Sin embargo, los de Cleveland desarrollaban una inflamación grave y fallecían meses antes que sus homólogos alojados en el Broad Institute de Cambridge (Massachusetts) o en el Jackson Laboratory de Bar Harbor (Maine). La diferencia, resultó, no estaba en sus genes. Estaba en su intestino.
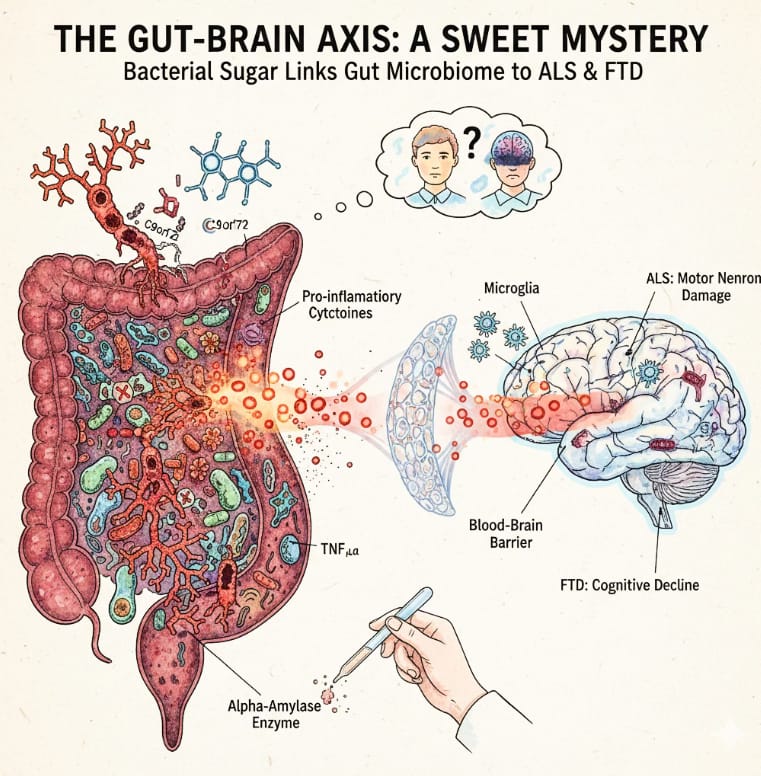
Burberry y su equipo han rastreado ahora el problema hasta algo aparentemente sencillo: un tipo de azúcar denominado glucógeno, producido por ciertas bacterias intestinales, que desencadena una cascada inmunitaria capaz de atravesar la barrera hematoencefálica y dañar el tejido neural. El hallazgo, publicado en Cell Reports(https://www.cell.com/cell-reports/fulltext/S2211-1247(25)01678-X) , podría transformar nuestra comprensión de dos de las afecciones neurológicas más devastadoras —la esclerosis lateral amiotrófica (ELA) y la demencia frontotemporal (DFT)— al señalar al microbioma intestinal como un desencadenante modificable de la enfermedad cerebral.
La ELA destruye las neuronas motoras, provocando una parálisis progresiva y, por lo general, la muerte en un plazo de dos a cinco años. Hasta el 15 % de las personas con ELA también desarrollan DFT, que deteriora la personalidad, la conducta y el lenguaje al afectar los lóbulos frontal y temporal del cerebro. Ambas patologías se sitúan en un espectro clínico compartido, y la causa genética más frecuente de las dos es una mutación en el gen C9ORF72, responsable de aproximadamente el 10 % de los casos. Sin embargo, persiste un enigma que ha inquietado a los investigadores durante años: no todas las personas portadoras de la mutación enferman. Algo ambiental debe actuar como desencadenante.
Ese algo, argumenta ahora el equipo de Burberry, es el glucógeno bacteriano.
Para resolver el caso, los investigadores adoptaron un enfoque metódico. Recolectaron materia fecal de ratones alojados en cuatro instituciones diferentes y la expusieron a células inmunitarias denominadas macrófagos. Las heces procedentes de entornos proinflamatorios (Cleveland y Harvard) provocaron una liberación mucho mayor de la citoquina TNF-alfa que las muestras de las instalaciones protectoras. Crucialmente, esta respuesta exagerada solo se observó en macrófagos carentes de C9orf72, lo que sugiere que el gen actúa normalmente como un freno de la inflamación desencadenada por microbios intestinales.
A continuación vino el trabajo de investigación para identificar qué bacterias eran responsables. El equipo evaluó 18 cepas bacterianas de forma individual y encontró 10 especies filogenéticamente diversas que incrementaban la liberación de citoquinas de manera dependiente de C9orf72. La secuenciación metatranscriptómica señaló un camino metabólico específico: la biosíntesis de glucógeno. Un algoritmo de aprendizaje automático entrenado con los datos clasificó las muestras fecales de entornos proinflamatorios frente a protectores con una precisión casi perfecta (área bajo la curva de 0,98). Cuando los investigadores trataron las bacterias problemáticas con alfa-amilasa —una enzima que degrada el glucógeno—, la señal inflamatoria disminuyó sustancialmente en cuatro de las cinco especies clave. El azúcar era el arma.
Bajo el microscopio electrónico se podía observar directamente. Las bacterias tratadas con alfa-amilasa aparecían vaciadas en comparación con los controles no tratados, con una densidad intracelular visiblemente reducida, coherente con el agotamiento de las reservas de glucógeno. Y no se trataba de cualquier glucógeno. El análisis estructural reveló que las formas inflamatorias presentaban cadenas de azúcar más cortas y ramificaciones más frecuentes que las variedades benignas: una arquitectura compacta que las hace más termoestable y, aparentemente, más irritante para el sistema inmunitario.
“Hemos descubierto que bacterias intestinales perjudiciales producen formas inflamatorias de glucógeno… y que estos azúcares bacterianos desencadenan respuestas inmunitarias que dañan el cerebro”, afirmó Burberry. El hallazgo explica un enigma que ha persistido durante décadas. ¿Por qué algunos portadores de la mutación C9ORF72 desarrollan ELA o DFT mientras otros viven sin verse afectados? La respuesta podría depender, en parte, de qué microbios han colonizado su intestino.
La evidencia más contundente provino de ratones libres de gérmenes. El equipo rederivado sus animales deficientes en C9orf72 en condiciones completamente estériles y luego introdujo selectivamente distintas comunidades bacterianas. Los ratones que permanecieron libres de gérmenes mostraron solo inflamación leve. Aquellos colonizados con bacterias protectoras del Jackson Laboratory evolucionaron razonablemente bien. Sin embargo, los ratones colonizados con una sola especie, Parabacteroides merdae, junto a una flora intestinal por lo demás benigna, desarrollaron un síndrome inflamatorio grave: esplenomegalia, monocitosis elevada, infiltración de linfocitos T en la médula espinal e inmunoglobulinas que atravesaban la barrera hematoencefálica hacia el cerebro. Con C9orf72 intacto, estas mismas exposiciones bacterianas causaron mucho menos daño.
A continuación, el equipo probó algo que suena casi demasiado sencillo. Tomaron ratones deficientes en C9orf72 alojados en condiciones convencionales que ya presentaban signos de enfermedad (mal rendimiento motor, marcadores sanguíneos elevados, alrededor de seis meses de edad) e iniciaron dosis orales diarias de alfa-amilasa. En el transcurso de 10 semanas, todos los ratones tratados sobrevivieron. Entre los controles tratados con vehículo, tres de seis machos fallecieron. La enzima también redujo el tamaño del bazo y atenuó una firma génica inflamatoria dependiente de C9orf72 en la microglía cerebral, redirigiendo esencialmente a estas células inmunitarias lejos de su preocupación por detritos bacterianos circulantes. La alfa-amilasa no lo corrigió todo: el rendimiento motor no mejoró y los marcadores sanguíneos permanecieron persistentemente elevados. Pero los animales tratados vivieron, y sus cerebros mostraron menor inflamación.
Cuando los investigadores examinaron muestras humanas, el patrón se mantuvo. Obtuvieron materia fecal de 23 pacientes con ELA o DFT y 12 controles sanos procedentes de centros en Estados Unidos e Italia. El glucógeno inflamatorio estaba presente en el intestino de alrededor del 70 % de los pacientes, pero solo en un tercio de los individuos sanos. En el grupo ELA/DFT, la señal sensible a alfa-amilasa fue estadísticamente significativa. Entre los controles sanos, no lo fue. Ocho de nueve muestras recolectadas en los primeros 18 meses tras el diagnóstico de ELA contenían glucógeno inflamatorio, lo que sugiere que el azúcar podría ser especialmente relevante en las fases tempranas de la enfermedad.
Los hallazgos son sugestivos más que definitivos, ya que la cohorte humana fue pequeña y el estudio no pudo determinar si el glucógeno inflamatorio precedía al inicio de la enfermedad. Burberry reconoce que el glucógeno bacteriano es probablemente uno entre varios factores ambientales que interactúan con genotipos predisponentes. Sin embargo, las implicaciones terapéuticas son tentadoras. Reducir los azúcares perjudiciales en el intestino “mejoró la salud cerebral y prolongó la supervivencia” en el modelo murino, según Rodriguez-Palacios. Ensayos clínicos podrían comenzar en el plazo de un año, indicó Burberry, para evaluar si la degradación del glucógeno en el intestino de pacientes con ELA y DFT podría ralentizar la progresión de la enfermedad.
Hay también una lección más amplia. Tendemos a concebir la neurodegeneración como un problema que comienza y termina en el cerebro. Pero el intestino alberga aproximadamente 100 billones de microorganismos, con los que interactúa el 70-80 % de las células inmunitarias del organismo. ¿Y si, para algunas personas portadoras de la predisposición genética a la ELA o la DFT, el momento crítico no fuera un fallo dentro de una neurona motora, sino una bacteria concreta en el colon activando una vía de biosíntesis de glucógeno? Es una idea inquietante, pero también, quizá, esperanzadora. No se pueden cambiar los genes. Pero tal vez, en el futuro, sí se puede modificar lo que las bacterias intestinales están produciendo.
Enlace al estudio: https://www.cell.com/cell-reports/fulltext/S2211-1247(25)01678-X