Dedicado a la memoria de Herland Vacadiez Busch, el mejor nefrólogo de Bolivia hasta que migró a otro planeta
Durante años, la patogenia de los síndromes nefróticos de inicio agudo, como la enfermedad de cambios mínimos y la glomeruloesclerosis focal y segmentaria (GEFS), ha carecido de una fisiopatogenia clara. Sin embargo, un déficit de nefrina, una proteína de superficie y señalización transmembrana del podocito,(1) ahora está en el centro de atención como un factor impulsor de la enfermedad de cambios mínimos y una forma de GEFS. (El podocito es una célula epitelial especializada del glomérulo). De hecho, los anticuerpos contra la nefrina que se inyectan en ratas causan proteinuria(2), y Weins y sus colegas informaron sobre autoanticuerpos específicos contra la nefrina en humanos con enfermedad de cambios mínimos en 2022.
Hengel et al.(4 )informan que 46 de 105 pacientes (44%) con enfermedad de cambios mínimos y 7 de 74 pacientes (9%) con FSGS primaria tenían autoanticuerpos antinefrina; entre los pacientes con enfermedad activa que no habían recibido tratamiento inmunosupresor, los porcentajes fueron del 69% y el 90%, respectivamente. Los autores también describen cómo los autoanticuerpos antinefrina pueden incitar la glomerulopatía. Es necesario comprender la estructura y la función del riñón y su tarea principal (la producción de orina) para apreciar plenamente las implicaciones de este estudio.
Filtración de la sangre y formación de la orina.
La sangre de un adulto sano medio se filtra a través de los aproximadamente 2 millones de glomérulos que hay en los riñones, ¡cada 5 minutos! .Por lo tanto, cada 24 horas, un ser humano adulto produce el equivalente a 10 galones de filtrado. En última instancia, la mayor parte de este filtrado se reabsorbe en los túbulos renales, lo que da como resultado la producción neta de uno o dos litros de orina, que, en personas sanas, está casi libre de proteínas y moléculas grandes, pero contiene sustancias de desecho más pequeñas (Figura 1).
=> Recibir por Whatsapp las noticias destacadas
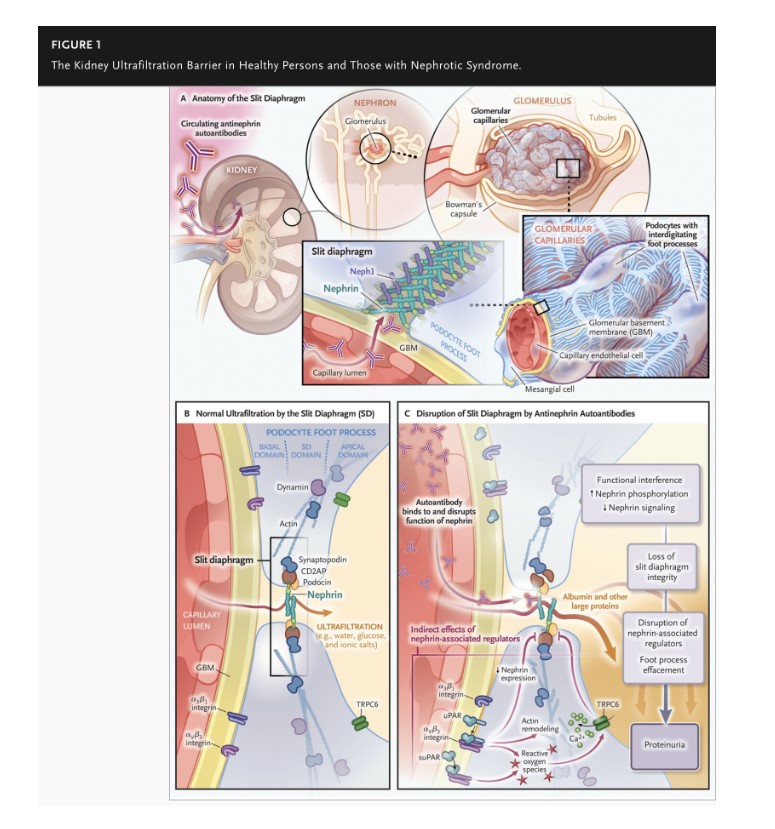
El deterioro del proceso de filtración supone una gran carga: aproximadamente el 12% de las personas con dicho deterioro acaban teniendo algún tipo de enfermedad renal, y la mayoría de las personas con enfermedad renal tienen glomérulos disfuncionales, que se caracterizan por una filtración reducida de los productos de desecho del torrente sanguíneo y una mayor pérdida de proteínas que, de otro modo, mantendrían la presión oncótica en la circulación.
¿Cómo filtra la sangre el riñón?
Dentro de cada glomérulo, la sangre se filtra a medida que se mueve bajo presión a través de capilares especializados, cuyas paredes están formadas por tres capas: una capa endotelial fenestrada que retiene glóbulos rojos y moléculas mayores de aproximadamente 6 a 8 nm, una membrana basal y una capa de podocitos(5). Cada podocito tiene muchos “procesos de pies” largos similares a tentáculos; en el riñón sano, el extremo plano (la placa de la suela) de cada proceso de pie está firmemente sellado a la membrana basal, unido a ella por moléculas de adhesión llamadas integrinas. El funcionamiento adecuado de este sello y el del diafragma de hendidura, una unión especializada entre los procesos de pie de los podocitos vecinos que está formada por proteínas estructurales (como la nefrina, que se extiende desde la superficie del podocito hasta el diafragma de hendidura), es fundamental para el proceso de filtración de la sangre. De hecho, las variantes patógenas en más de 60 genes que codifican proteínas de los podocitos (varios de los cuales son componentes del diafragma de hendidura) pueden causar enfermedad renal(6). Por ejemplo, las variantes en NPHS1, que codifica la nefrina, causan una forma congénita de síndrome nefrótico(3), y las variantes en TRPC6, un canal iónico transmembrana al que se une la nefrina en cis, causan FSGS(7).
¿Qué causa la lesión del podocito?
Las variantes genéticas patógenas que provocan cambios en las proteínas estructurales representan una fuente de lesión. Sin embargo, el aparato de filtrado del glomérulo tiene propiedades mecánicas y de señalización que son sensibles a las proteínas inflamatorias circulantes, las toxinas y los anticuerpos. La agresión inflamatoria puede ser causada por una infección viral. En este caso, las moléculas de integrina en la placa base del proceso del pie del podocito pueden ser activadas por una molécula mensajera del sistema inmunitario innato, el receptor soluble del activador del plasminógeno de la uroquinasa (suPAR), lo que conduce a cambios en la expresión de la nefrina, un acortamiento del diafragma de hendidura y, finalmente, proteinuria(8).
Los cambios en el diafragma de hendidura que son inducidos por autoanticuerpos antinefrina también comprometen la filtración glomerular. El hecho de que los niveles de estos autoanticuerpos antinefrina aumenten y disminuyan en asociación con la actividad de la enfermedad, en particular en pacientes con enfermedad de cambios mínimos, implica que la focalización de estos anticuerpos o las células B que los producen podría inducir la remisión. De hecho, Hengel et al. informan de una remisión en dos pacientes, uno con enfermedad de cambios mínimos y el otro con FSGS, que recibieron tratamiento con rituximab, que elimina las células B.
El hallazgo de autoanticuerpos contra diferentes componentes del mecanismo de filtración glomerular puede explicar otras afecciones, como el síndrome de Goodpasture (causado por autoanticuerpos contra un tipo de colágeno que se encuentra en la membrana basal glomerular) y la nefropatía membranosa primaria (causada por autoanticuerpos contra proteínas expresadas por el podocito). Aún está por dilucidar cómo exactamente los autoanticuerpos interfieren con los componentes del aparato de filtración(6).
Dicho esto, Hengel et al. han proporcionado cierta información. Modelaron la enfermedad renal podocitopática inmunizando ratones con el ectodominio de la nefrina de ratón, lo que llevó al rápido desarrollo del síndrome nefrótico con características de la enfermedad de cambios mínimos. Además, el anticuerpo llevó a una redistribución de la nefrina desde la superficie celular al citoplasma del podocito. Hengel et al. también se observó un aumento de la fosforilación de la nefrina en un aminoácido específico de la nefrina (tirosina en la posición 1176); la fosforilación en este sitio en particular está asociada con la endocitosis de la nefrina y la reorganización del citoesqueleto. No hubo signos de activación inmunitaria aparte de la respuesta de anticuerpos.
¿Qué sigue?
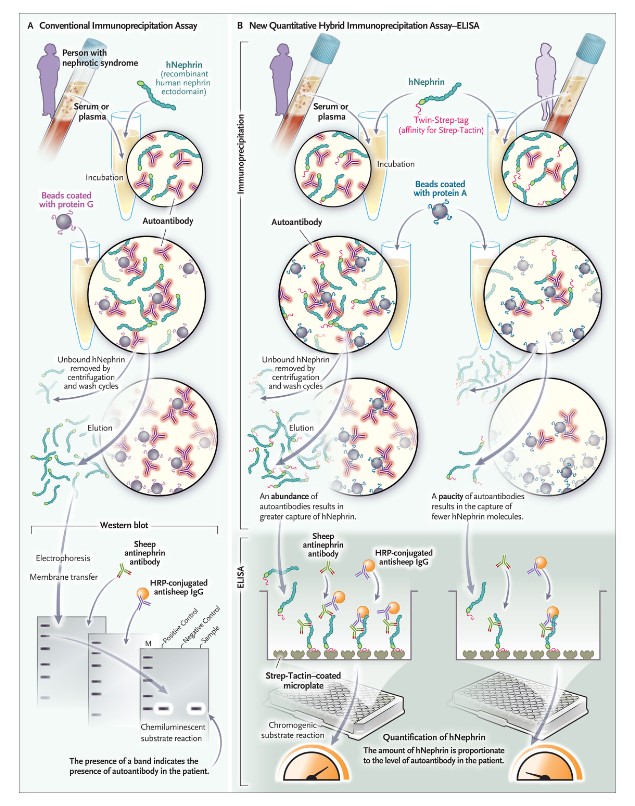
Los hallazgos informados por Hengel et al. y otros(9,10) apuntan hacia un objetivo clínico: la incorporación de una evaluación cuantitativa, rutinaria y confiable de los autoanticuerpos como herramientas de diagnóstico. Hengel et al. desarrollaron un ensayo inmunoabsorbente ligado a enzimas cuantitativo (Figura 2) para seguir el aumento y la disminución de los niveles de autoanticuerpos. Estos hallazgos también incentivan la investigación sobre cómo se pueden mitigar mejor los efectos de los autoanticuerpos antinefrina. Hoy en día,
Figura 2.
Dicha mitigación se puede lograr ampliamente a través del intercambio de plasma o el uso de medicamentos basados en anticuerpos que eliminan las células B, como el rituximab. Se pueden desarrollar terapias más específicas que reduzcan, bloqueen o eliminen los autoanticuerpos antinefrina. En última instancia, el tratamiento guiado por la evaluación de los autoanticuerpos requeriría una comparación formal con el tratamiento clínico actual.
Referencias Bibliográficas
1.
Kestilä M, Lenkkeri U, Männikkö M, et al. Positionally cloned gene for a novel glomerular protein — nephrin — is mutated in congenital nephrotic syndrome. Mol Cell 1998;1:575-582.
Go to Citation
2.
Topham PS, Kawachi H, Haydar SA, et al. Nephritogenic mAb 5-1-6 is directed at the extracellular domain of rat nephrin. J Clin Invest 1999;104:1559-1566.
Go to Citation
3.
Watts AJB, Keller KH, Lerner G, et al. Discovery of autoantibodies targeting nephrin in minimal change disease supports a novel autoimmune etiology. J Am Soc Nephrol 2022;33:238-252.
4.
Hengel FE, Dehde S, Lassé M, et al. Autoantibodies targeting nephrin in podocytopathies. N Engl J Med 2024;391:422-433.
5.
Benzing T, Salant D. Insights into glomerular filtration and albuminuria. N Engl J Med 2021;384:1437-1446.
6.
Meliambro K, He JC, Campbell KN. Podocyte-targeted therapies — progress and future directions. Nat Rev Nephrol 2024 May 9 (Epub ahead of print).
7.
Reiser J, Polu KR, Möller CC, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 2005;37:739-744.
8.
Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem 2001;276:41543-41546.
Go to Citation
9.
Wei C, Datta PK, Siegerist F, et al. SuPAR mediates viral response proteinuria by rapidly changing podocyte function. Nat Commun 2023;14:4414-4414.
Go to Citation
10.
Shirai Y, Miura K, Ishizuka K, et al. A multi-institutional study found a possible role of anti-nephrin antibodies in post-transplant focal segmental glomerulosclerosis recurrence. Kidney Int 2024;105:608-617.
Pruebas para detectar autoanticuerpos antinephrin