Resumen
Durante más de un siglo, los médicos han buscado formas de reducir farmacológicamente el exceso de grasa corporal. La situación finalmente ha cambiado con los recientes avances en agonistas diseñados bioquímicamente para el receptor del péptido similar al glucagón-1 (GLP-1) y su uso en poliagonistas basados en GLP-1. Estos poliagonistas reducen el peso corporal a través de la farmacología complementaria al incorporar los receptores de glucagón y/o el polipéptido insulinotrópico dependiente de la glucosa (GIP). En sus formas más avanzadas, los poliagonistas de hormonas intestinales logran una reducción de peso sin precedentes de hasta ∼20%–30%, ofreciendo una alternativa farmacológica a la cirugía bariátrica. Junto con los efectos favorables sobre la glucemia, el hígado graso y la enfermedad renal, también ofrecen efectos beneficiosos sobre el sistema cardiovascular y el tejido adiposo. Por lo tanto, estas nuevas intervenciones son muy prometedoras para el futuro de los medicamentos contra la obesidad.
Introducción
La obesidad, caracterizada por el exceso de grasa corporal, constituye un factor de riesgo importante para el desarrollo de diabetes tipo 2 (DT2), dislipidemia, enfermedad cardiometabólica, cáncer y mortalidad general. Además, aumenta las complicaciones asociadas con enfermedades infecciosas, como lo ejemplifica la enfermedad por coronavirus 2019 (COVID-19). Entre 1975 y 2014, las tasas mundiales de obesidad aumentaron de 105 a 641 millones de adultos (4% a 13% de la población total, respectivamente). Se estima que la obesidad mundial seguirá aumentando hasta mil millones de adultos en 2030, independientemente del género, la geografía o los estilos de vida rurales y urbanos. Las modificaciones del estilo de vida, con un mayor ejercicio físico y/o una ingesta calórica reducida, son características distintivas de cualquier intervención exitosa para perder peso. Sin embargo, a pesar de una pérdida de peso apreciable de aproximadamente el 5% al 8% en el corto plazo, estas intervenciones en el estilo de vida solo tienen un potencial limitado para una reducción de peso sostenida, en particular cuando se utilizan como una medida independiente. Un metanálisis reciente demuestra que aproximadamente el 56% de la pérdida de peso corporal, lograda mediante una intervención en el estilo de vida, se recupera en un plazo de 2 años, y aproximadamente el 79% se recupera después de 5 años. El principal desafío en el manejo de la obesidad es el impulso intrínseco del sistema de conservar energía para defender el mayor peso corporal. Por lo tanto, una reducción en la ingesta calórica suele ir acompañada de una disminución en el gasto energético, junto con una mayor sensibilidad a los factores que estimulan la ingesta de alimentos. Combinadas, estas respuestas obstaculizan la pérdida de peso y promueven la recuperación de peso. Hasta hace poco, la cirugía bariátrica había sido el tratamiento más eficaz para mantener una reducción en el peso corporal y, como tal, es el punto de referencia actual para los medicamentos contra la obesidad.
=> Recibir por Whatsapp las noticias destacadas
La regulación del peso corporal está orquestada principalmente por el cerebro y el tejido adiposo (Figura 1), que integran constantemente información relacionada con el estado energético del cuerpo para ajustar la ingesta de alimentos, la saciedad y el equilibrio energético. Las hormonas notables implicadas en este eje de comunicación intestino-cerebro-grasa incluyen, entre muchas otras, las adipocinas leptina y adiponectina, la hormona secretada por el hígado factor de crecimiento de fibroblastos 21 (FGF21), la hormona derivada de las células α pancreáticas glucagón, los péptidos del sistema gastrointestinal grelina, péptido YY (PYY) y colecistoquinina (CCK), además de las incretinas péptido similar al glucagón-1 (GLP-1) y polipéptido insulinotrópico dependiente de la glucosa (GIP).
El glucagón, conocido tradicionalmente por su capacidad para contrarrestar la hipoglucemia, ha estado implicado en la patogénesis de la diabetes tipo 1 y la diabetes tipo 2 . El glucagón actúa a través del receptor de glucagón (GCGR), y las alteraciones en la señalización de GCGR pueden tener efectos profundos en el metabolismo de la glucosa. Si bien la falta de señalización de GCGR puede normalizar la glucemia en condiciones de deficiencia de insulina, este efecto depende de la presencia de insulina residual. Curiosamente, el glucagón tiene una biología pleiotrópica que se extiende más allá de su papel en el control de la glucemia. En particular, los estudios preclínicos han revelado que el glucagón puede aprovecharse para obtener beneficios metabólicos, como la pérdida de peso corporal en el contexto de la obesidad. Se ha demostrado que la administración de glucagón a ratas promueve un balance energético negativo al aumentar el consumo de oxígeno, un efecto que luego se atribuyó a un aumento de la termogénesis sin escalofríos. Además, se informó que una mejor señalización de GCGR reduce eficazmente el peso corporal mediante la inhibición de la ingesta de alimentos y la estimulación del gasto energético, y modula aún más el metabolismo lipídico al impulsar la lipólisis e inhibir la lipogénesis. El glucagón también tiene la capacidad de inhibir la motilidad gástrica y promover la filtración glomerular renal, con efectos notables evidentes en el sistema cardiovascular para aumentar la frecuencia cardíaca, la contractilidad cardíaca y el gasto cardíaco (Figura 1). En conjunto, esto respalda la perspectiva de que el agonismo de GCGR se puede emplear como una opción viable para el tratamiento de enfermedades metabólicas asociadas con la obesidad, particularmente cuando se utiliza como complemento de terapias capaces de restringir las limitaciones glucémicas y cardiovasculares agudas del glucagón.
Descubrimiento de las hormonas incretinas y de la acción metabólica del glucagón.
Desde hace tiempo se reconoce que el intestino es un actor clave en la regulación del metabolismo de la glucosa. En 1906, se descubrió que los extractos de la mucosa intestinal disminuyen la glucosuria en sujetos con diabetes tipo 2 (DT2)y que la excursión de la glucosa en sangre es mucho mayor cuando la glucosa pasa a través del intestino en relación con la infusión intravenosa. Atribuido a una mayor secreción de insulina estimulada por la glucosa, esto señaló al intestino como el origen de las hormonas insulinotrópicas, que posteriormente se identificaron como el polipéptido insulinotrópico dependiente de la glucosa (GIP) en 1973 y el péptido similar al glucagón-1 (GLP-1) en 1987. El GIP se denominó inicialmente polipéptido inhibidor gástrico debido a su inhibición de la secreción de ácido gástrico en dosis suprafisiológicas.
En 1980, un estudio encontró que el efecto insulinotrópico de la glucosa ingerida por vía oral disminuyó en sujetos con enfermedad de Crohn que se sometieron a resección ileal. Dado que el deterioro de la secreción de ácido gástrico en pacientes con enfermedad de Crohn se asoció con una disminución de la secreción de ácido gástrico, se descubrió que la glucosa ingerida por vía oral disminuyó en pacientes con enfermedad de Crohn que se sometieron a resección ileal. Aunque la respuesta de la incretina no estaba relacionada con la secreción de GIP inducida por glucosa, se planteó la hipótesis de que era probable que el intestino inferior albergara una hormona insulinotrópica adicional similar a GIP. En 1983, Creutzfeldt informó además un efecto de la incretina parcialmente conservado cuando se neutralizó el GIP de los extractos intestinales de rata, mientras que Habener descubrió que el ADNc del preproglucagón del rape codifica una nueva secuencia con una homología de secuencia considerable con el glucagón. Posteriormente, se identificaron dos péptidos similares al glucagón, GLP-1 y GLP-2, en las secuencias de proglucagón de hámster, humano, y rata. Como se sabía que tanto el glucagón como el GIP estimulaban la secreción de insulina, se planteó la hipótesis de que los péptidos recientemente identificados también podrían promover la secreción de insulina. Se demostró que el proglucagón produce varias formas más cortas de GLP-1, y aunque ni el GLP-2 ni el GLP-1 de longitud completa estimularon la secreción de insulina, dos formas truncadas N-terminales del péptido, GLP-1 (7-37) y GLP-1 (7-36NH2), mostraron una acción insulinotrópica en páncreas perfundidos de ratas y cerdos, además de la línea celular β de insulinoma de rata RIN 1046. La identificación de diferentes formas del péptido, así como los estudios de correlación sobre la actividad insulinotrópica de las diferentes formas de GLP-1 en el páncreas de rata perfundido aislado, por Mojsov, contribuyeron a establecer el GLP-1 como una hormona incretina, poco después de ser confirmado en humanos. GIP y Por lo tanto, se estableció que el GLP-1 era la hormona incretina predominante. Más tarde, también se descubrió que el GLP-1 afectaba el metabolismo de la glucosa a través de la inhibición de la secreción de glucagón y la motilidad gástrica. La relevancia de estos efectos no insulinotrópicos del GLP-1 se reveló en estudios de fijación de glucosa en un entorno de diabetes tipo 2, donde la inhibición de la secreción de glucagón demostró ser tan importante en el control glucémico como la secreción mejorada de insulina.
El glucagón se descubrió en 1923 a partir de homogeneizados pancreáticos durante el refinamiento de la purificación de la insulina. Cinco décadas después, durante las cuales el glucagón se caracterizó químicamente y se estableció biológicamente como una hormona contrarreguladora de la insulina, todavía se le otorgaba poco o ningún valor farmacológico más allá de su capacidad para rescatar la hipoglucemia aguda. De hecho, se propuso que el glucagón fuera un elemento causal en la patogénesis de la diabetes. Aparentemente consistente con esto fue la observación de que el receptor de glucagón en os ratones deficientes en (Gcgr) están protegidos de la diabetes inducida por estreptozotocina. Sin embargo, estudios posteriores revelaron que la insulina residual es fundamental para la normalización de la glucemia en ratones deficientes en Gcgr tratados con estreptozotocina,y, además, la diabetes tipo 1 (DT1) se desarrolla rápidamente en animales experimentales después de la extirpación quirúrgica del páncreas. En conjunto, esto indicó que la DT1 se origina por la falta de insulina en lugar del exceso de glucagón. No obstante, la búsqueda del antagonismo del glucagón como posible terapia antihiperglucémica evolucionó y fue apoyada por la casi normalización de la glucemia después de la inhibición farmacológica, o genética de la señalización de GCGR en roedores deficientes en insulina. Los efectos glucémicos beneficiosos de la inhibición de la señal de GCGR también se han informado a partir de estudios clínicos en sujetos con diabetes tipo 2. Aunque el antagonismo de GCGR en ratones a menudo resulta en hiperplasia de células α pancreáticas, no se observa tal efecto en primates no humanos. Por otro lado, existen preocupaciones persistentes de que el antagonismo de GCGR pueda aumentar los niveles de colesterol total y de lipoproteínas de baja densidad. Irónicamente, mientras que el antagonismo de GCGR ha caído en desgracia actualmente a medida que la obesidad emergió como una característica de confusión prominente de la diabetes tipo 2, la enfermedad del hígado graso y la aterosclerosis, hemos sido testigos de un renacimiento reciente en la utilización del agonismo de GCGR, predominantemente en formulaciones unimoleculares con agonismo de GLP-1R, para el tratamiento de exactamente estas enfermedades metabólicas.
Se descubrió inicialmente que el GIP derivado de la hormona intestinal, secretado por las células K enteroendocrinas en el intestino superior, en las mucosas del duodeno y el yeyuno, desempeñaba un papel integral en la respuesta del cuerpo a la ingesta de glucosa . El GIP ejerce un papel significativo en el flujo sanguíneo del tejido adiposo y, en condiciones de hiperinsulinemia, promueve la deposición de lípidos en los adipocitos al estimular la lipoproteína lipasa. La hormona intestinal también potencia la captación de glucosa inducida por insulina, lo que conduce a un aumento en la conversión de lípidos a partir de la glucosa. Sin embargo, in vivo, se demostró que el GIP promueve la lipólisis en condiciones de normo o hipoinsulinemia, y los ratones que sobreexpresan GIP exhiben una menor masa grasa cuando se alimentan con una dieta alta en grasas. Los estudios preclínicos revelaron además que la activación quimiogenética de las neuronas GIPR en el hipotálamo o en el rombencéfalo reduce la ingesta de alimentos (Figura 1) y que el agonismo central de GIPR mejora el efecto emético del agonismo del receptor GLP-1 (GLP-1R) y estimula la pérdida de peso a través de la inhibición de la ingesta de alimentos. De hecho, la pérdida de peso impulsada por GIP La incretina GLP-1 se potencia sinérgicamente mediante el agonismo adjunto de GLP-1R, y, aunque la pérdida de peso mediada por GIP se conserva por completo en ratones deficientes en Glp1r,el GIP no altera la ingesta de alimentos ni el peso corporal en ratones que albergan una deleción de Gipr en el sistema nervioso central (SNC) o, más específicamente, en las neuronas γ-aminobutíricas (GABA)érgicas.
La otra incretina, GLP-1, secretada por las células L enteroendocrinas en el íleon y las mucosas colónicas del intestino grueso, se identificó por su papel en la estimulación de la insulina, la inhibición de la secreción de glucagón y la motilidad gástrica, ambos efectos esenciales en la regulación de la homeostasis de la glucosa. De hecho, los estudios han demostrado que la inhibición de la secreción de glucagón por GLP-1 es tan importante como la secreción mejorada de insulina para controlar los niveles de glucosa en la diabetes tipo 2. Más allá de su papel en el metabolismo de la glucosa, se sabe que GLP-1 posee efectos cardio y neuroprotectores, reduce la apoptosis celular y la inflamación, y modula el comportamiento de recompensa y la palatabilidad (Figura 1). Además, GLP-1 ejerce un efecto significativo sobre el peso corporal al inhibir la ingesta de alimentos a través de mecanismos mediados centralmente.7 Este último aspecto despertó un enorme interés en GLP-1 y, junto con GIP, los colocó a ambos en una trayectoria como candidatos prometedores para su uso en el tratamiento de la obesidad.
La acción fisiológica del eje GLP-1/GIP convirtió a estas hormonas intestinales en objetivos medicinales atractivos para tratar la diabetes tipo 2 y, posteriormente, la obesidad. En particular, el agonismo de GLP-1R no solo surgió como una herramienta poderosa en el tratamiento de la diabetes tipo 2 y el exceso de adiposidad, sino que también mostró efectos favorables en el sistema cardiovascular y las enfermedades neurodegenerativas. Esto resalta que los agonistas de GLP-1R tienen un perfil de acción apreciable fuera de sus objetivos originales en el páncreas. El cerebro no depende de un solo factor para regular el metabolismo energético; más bien, integra una variedad de señales independientes para ajustar la ingesta y el gasto de energía. De manera similar, el adipocito está en la primera línea de las reservas calóricas, el influjo calórico y el gasto de energía y, como tal, está bien posicionado para orquestar respuestas sistémicas a través de múltiples señales.
En consecuencia, se esperaría que una farmacoterapia que involucre múltiples señales metabólicas clave logre una mayor pérdida de peso en relación con un fármaco que se dirige solo a una vía de señalización metabólicamente relevante. En consonancia con este modelo, existe una batería de estudios preclínicos y clínicos que demuestran que la pérdida de peso inducida por el agonismo del receptor de GLP-1 se potencia cuando se administra de forma conjunta con glucagón, GIP, amilina, CCK o FGF21, lo que se puede lograr como coterapia o en una formulación poliamolecular de poliagonista. Estas polifarmacoterapias están diseñadas para permitir una dosificación individual submáxima en cada receptor diana y, por lo tanto, no solo pueden optimizar la pérdida de peso a través de la farmacología complementaria en varios receptores independientes, sino que también mejoran la tolerabilidad y reducen la probabilidad de taquifilaxia. La reciente aparición de poliagonistas unimoleculares constituye una vanguardia en los fármacos de próxima generación para el tratamiento de enfermedades metabólicas, como la obesidad. Los fármacos que poseen agonismo de GLP-1R con farmacología complementaria a través del GIPR han producido una eficacia sorprendente en la pérdida de peso, concomitante con un excelente perfil de seguridad y resultados metabólicos superiores en relación con los mejores agonistas de GLP-1R de su clase. La farmacología complementaria a través del receptor de glucagón, ya sea en combinación unimolecular con GLP-1R o agregada al coagonismo GLP-1R/GIPR, está estableciendo actualmente un nuevo punto de referencia. Estos agentes de última generación no solo aceleran una mayor pérdida de peso, sino que también abordan las comorbilidades asociadas con la obesidad, como la enfermedad del hígado graso, la enfermedad cardiovascular y la dislipidemia. En este artículo, analizamos el turbulento camino y las controversias que se han vivido durante décadas hasta llegar a la integración de GLP-1, GIP y glucagón como socios sinérgicos en terapias unimoleculares de vanguardia basadas en incretinas, destacando los logros clínicos instrumentales hasta la fecha.
El desarrollo de agonistas de GLP-1R en el tratamiento de la diabetes tipo 2 y la obesidad.
La identificación de GLP-1 como una hormona insulinotrópica, junto con la demostración de que la incretina tiene efectos beneficiosos mucho más allá de su acción sobre el páncreas (Figura 1), generó un gran interés por explorar su potencial farmacológico para el tratamiento de la diabetes tipo 2 y la obesidad. Sin embargo, el uso farmacológico del GLP-1 bioactivo nativo está limitado por una vida media extremadamente corta (∼2–3 min), resultante de la rápida eliminación renal y degradación proteolítica por la dipeptidil peptidasa-4 (DPP-4) y la endopeptidasa neutra . Se estima que tan solo el 10% del GLP-1 activo llega a la circulación general, y solo una mera fracción de este llega al cerebro. No obstante, la infusión continua de GLP-1, o la administración repetida de inhibidores de DPP-4, mejora el metabolismo de la glucosa en sujetos con DT2.
Las limitaciones farmacocinéticas del GLP-1 nativo se han mejorado mediante una variedad de modificaciones químicas, que sirven para mejorar la farmacología de la hormona a través de una mayor estabilidad molecular, una concentración plasmática mejorada y un aclaramiento renal retardado. Varios análogos selectivos del GLP-1 han recibido aprobación regulatoria en la última década e incluyen formulaciones adecuadas para inyecciones subcutáneas dos veces al día (exenatida), diarias (liraglutida, lixisenatida) y semanales (exenatida de liberación prolongada, albiglutida, dulaglutida y semaglutida). Más recientemente, se ha presentado el desarrollo de una forma de semaglutida de administración oral diaria como una alternativa prometedora a las inyecciones semanales. Si bien estos péptidos son altamente eficaces para el tratamiento de la diabetes tipo 2, como clase de fármaco, todos ellos demuestran efectos adversos gastrointestinales transitorios dependientes de la dosis, como náuseas y vómitos, que requieren una escalada de dosis cuidadosamente orquestada para alcanzar los efectos máximos.
La liraglutida (3 mg) fue el primer agonista del receptor del GLP-1 registrado para el tratamiento de la obesidad. Este agonista del receptor de GLP-1 se aprobó inicialmente para el tratamiento de la obesidad en adultos y, posteriormente, para la obesidad en niños y adolescentes. En sujetos con obesidad sin diabetes tipo 2, tras 1 año de tratamiento con liraglutida, se logró una pérdida de peso corregida con placebo de aproximadamente el 5,2 %, y aproximadamente un tercio de los sujetos alcanzaron una pérdida de peso de >10 %. En este estudio, la reducción del peso corporal inducida por liraglutida se asoció con un mejor control de la glucosa, una disminución de la presión arterial sistólica y diastólica (−2,8 y −0,9 mmHg respecto a los controles tratados con placebo), junto con una mejora de los perfiles de lípidos y colesterol, aunque con un ligero aumento de la frecuencia cardíaca de 2,4 latidos/min en comparación con los controles tratados con placebo. En 2021, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó la semaglutida (2,4 mg) una vez a la semana (OW) para el tratamiento de la obesidad. Después de 68 semanas de tratamiento, la semaglutida redujo de forma impresionante el peso corporal en individuos obesos no diabéticos en un 14,9%, en relación con el 2,4% en los controles tratados con placebo. Por el contrario, la semaglutida fue menos eficaz en sujetos con obesidad y diabetes tipo 2, con una pérdida de peso corregida por placebo de solo el 6,2% con 68 semanas de tratamiento.
Sin embargo, estos resultados resaltan la capacidad de sentar precedentes de los agonistas del receptor de GLP-1, cuando se titulan adecuadamente las dosis, para reducir significativamente el peso corporal. Además, sientan las bases para lograr una mayor pérdida de peso que la mayoría de los individuos obesos requieren con medios farmacológicos y, como tal, plantean la pregunta crítica de si se puede lograr una pérdida de peso adicional con farmacología complementaria.
Agonistas multirreceptores de hormonas intestinales para el tratamiento de la diabetes tipo 2 y la obesidad.
Los agonistas multirreceptores unimoleculares que emplean varias vías de señalización independientes están surgiendo como los mejores fármacos de su clase para el control de la glucemia y la pérdida de peso. La naturaleza pleiotrópica de la biología del glucagón, junto con un renovado interés en la farmacología del GIP, ha despertado un gran interés en explorar su uso terapéutico en formulaciones unimoleculares con agonismo del receptor de GLP-1 para tratar la obesidad y la diabetes. El objetivo ha sido aumentar la magnitud de la pérdida de peso posible en una amplia comunidad de personas con obesidad, sin imponer las limitaciones de seguridad que naturalmente residen en el agonismo del receptor de GLP-1. En particular, en sujetos con obesidad y diabetes tipo 2 persistente, la pérdida de peso impulsada por el receptor de GLP-1 todavía se estabiliza en el rango de un solo dígito, y, como tal, la eficacia mejorada de la farmacología complementaria para acelerar aún más la pérdida de peso, al mismo tiempo que se abordan de manera ideal las comorbilidades vinculadas con la obesidad, sigue siendo el objetivo principal. Se han explorado múltiples combinaciones de hormonas intestinales de manera preclínica, y un número apreciable ha avanzado a estudios clínicos, y los péptidos unimoleculares que poseen diversos grados de actividad del receptor de GLP-1, GIPR y GCGR constituyen el conjunto de candidatos a fármacos clínicamente más maduro. Aquí, analizaremos los estudios preclínicos y clínicos en coagonismo del receptor de GLP-1 con GIP o glucagón, así como los triagonistas completamente integrados.
Coagonistas de GLP-1R/GCGR
En los últimos años, ha surgido una mayor apreciación de la biología no pancreática del glucagón, específicamente basada en su papel como hormona reguladora del peso en el equilibrio energético y la saciedad. En roedores obesos inducidos por la dieta, la administración de agonistas de GCGR impulsa la pérdida de peso a través de una reducción en la ingesta de alimentos, una inducción de la utilización de lípidos a través de la termogénesis de la grasa parda, junto con un aumento en el gasto de energía de todo el cuerpo; el último efecto se atribuye en parte a la estimulación del FGF21 secretado por el hígado y la regulación positiva transcripcional del receptor nuclear hepático activado por ácidos biliares, el receptor farnesoide X. Esta regulación parcial en el gasto de energía sugiere que podría haber lugar para factores y mecanismos adicionales por los cuales la señalización mejorada de GCGR media el equilibrio energético. Curiosamente, el tratamiento de ratones obesos inducidos por la dieta con un agonista de GCGR reveló que el gasto de energía estimulado por glucagón y la pérdida de peso también pueden ser impulsados por el catabolismo de aminoácidos hepáticos y, por lo tanto, una respuesta sistémica a la hipoaminoacidemia. Además del hígado, los estudios preclínicos sugieren además un papel para el agonismo de GCGR en el riñón. El GCGR se regula negativamente de forma potente durante la enfermedad renal crónica, y la falta de señalización de glucagón en el riñón hace que el tejido sea susceptible a la fibrosis, la inflamación, el estrés oxidativo y la acumulación de lípidos. Esto indica que algunos de los beneficios cardiorrenales observados para los coagonistas pueden ejercer sus efectos beneficiosos directamente a través de un eje de señalización riñón-GCGR.
Junto con una homología de secuencia apreciable (∽50%) con las hormonas incretinas, los efectos no glucémicos del glucagón hacen que el péptido sea un candidato atractivo para una unión unimolecular con GLP-1 y GIP. Es importante destacar que los beneficios de dicha polifarmacoterapia se basan en el supuesto no solo de que estos agentes reforzarían la eficacia de la pérdida de peso a través de una farmacología complementaria en cada receptor objetivo, sino también de que los efectos glucémicos y cardiovasculares positivos de las incretinas limitarían cualquier efecto potencialmente perjudicial que pueda o no residir en el agonismo del GCGR. Los péptidos unimoleculares de GLP-1R y GCGR fueron los primeros coagonistas intencionales que surgieron, buscando amalgamar el aumento mediado por glucagón en el gasto de energía con la acción anoréxica de GLP-1R para promover la pérdida de peso, mientras que empleaban la acción antidiabética de GLP-1 para minimizar el riesgo diabetogénico del agonismo del receptor de glucagón sin oposición.
Una vez demostrado que la potencia total en cada uno de los dos receptores podía ensamblarse químicamente en un solo péptido quimérico de tamaño comparable a cada hormona nativa, los estudios en modelos animales de obesidad, utilizando coagonistas GLP-1R/GCGR de primera generación, dieron como resultado una pérdida de peso superior, una mayor eficacia para reducir la glucosa y una reducción en la ingesta de alimentos en comparación con los agonistas selectivos de GLP-1R (Figura 2). Estos informes preclínicos iniciales promovieron una avalancha de interés farmacéutico que validó estas observaciones iniciales hasta un punto en el que numerosos péptidos GLP-1R/GCGR han progresado desde el laboratorio hasta el desarrollo clínico, y los más caracterizados se describen a continuación.
El primer coagonista de GLP-1R/GCGR que surgió.
El primer coagonista de GLP-1R/GCGR evaluado preclínicamente se basó en la secuencia de glucagón, en la que se introdujeron gradualmente residuos de aminoácidos de GLP-1 y GIP para lograr una actividad equilibrada en ambos receptores diana. El péptido protegido con DPP-4 transportaba un polietilenglicol de 40 kDa para retrasar la depuración renal y, después de una dosificación semanal en ratones obesos inducidos por la dieta, el péptido redujo el peso corporal y la masa grasa en aproximadamente un 25,8 %, logrando estos efectos a través de la acción anoréxica sinérgica en ambos receptores diana, con efectos termogénicos y lipolíticos complementarios obtenidos a través del agonismo de GCGR. El SAR425899, desarrollado más recientemente, emplea elementos de glucagón en la secuencia de exendina-4 para proporcionar agonismo de GCGR mientras se mantiene la activación de GLP-1R. Se demostró que SAR425899 reduce eficazmente el peso corporal y la grasa En ratones obesos, se estimulan efectos glucémicos robustos en ratones db/db diabéticos deficientes en receptores de leptina y, notablemente, se reduce la ingesta calórica total y se aumenta el gasto energético en primates no humanos obesos y diabéticos. En un estudio de fase 2 compuesto por sujetos obesos con diabetes tipo 2, SAR425899 superó a liraglutida después de 26 semanas de tratamiento para producir mayores mejoras en el control glucémico posprandial, junto con una capacidad de respuesta superior de las células β pancreáticas y una mayor sensibilidad a la insulina. En un estudio de fase 1b más reciente en sujetos con sobrepeso y obesos, el tratamiento con SAR425899 durante 19 días redujo el peso corporal y aumentó la oxidación de lípidos en relación con los sujetos que recibieron una dieta restringida en calorías, y dichos efectos fueron notablemente consistentes con la capacidad termogénica del glucagón para mejorar el gasto energético. Este fue el primer ensayo en humanos que demostró que el coagonismo GLP-1R/GCGR podría ser una opción adecuada para el control del peso. Mantenimiento de la pérdida de peso y validación clínica adicional de que el agonismo del glucagón podría contribuir significativamente a la regulación del equilibrio energético en un entorno obeso (Figura 3). No obstante, el desarrollo clínico de SAR425899 finalmente se interrumpió debido a efectos adversos, principalmente náuseas y vómitos.
Mazdutide
Mazdutide (también denominado IBI362, oxintomodulina 3 [OXM-3] o LY3305677) es un coagonista sintético monocatenario de GLP-1R/GCGR análogo al OXM de los mamíferos y modificado con una cadena lateral de acilo graso para extender su vida media circulante. En ratones obesos inducidos por la dieta, se informó que mazdutide redujo el peso corporal, mejoró el control glucémico y aumentó el gasto de energía, con una acción parcialmente conservada evidente en ratones que albergaban una deleción de Glp1r o Gcgr. El péptido redujo aún más la ingesta de alimentos y mejoró la tolerancia a la glucosa tanto en ratones obesos inducidos por la dieta como en ratones diabéticos inducidos por estreptozotocina. Los beneficios metabólicos adicionales del tratamiento con mazdutide incluyen, entre otros, aumentar los niveles sistémicos de FGF21 y reducir los niveles plasmáticos de triglicéridos. Estas propiedades hacen que mazdutide sea un fármaco altamente eficaz, coagonista para tratar la disfunción metabólica asociada con la obesidad, particularmente a través de la estimulación indirecta de la acción de FGF21, uno de los FGF endocrinos clave conocidos por su capacidad para reducir eficazmente los niveles sistémicos de triglicéridos en humanos. Un estudio reciente de fase 2, compuesto por 248 sujetos chinos con sobrepeso y obesidad, documentó una reducción del peso corporal de ∼11,3% desde el inicio después de 24 semanas de tratamiento con mazdutida en la dosis más alta. Se demostró además que el péptido reduce la presión arterial; reduce los niveles de lípidos, ácido úrico en sangre y transaminasas; y alivia la acumulación de lípidos hepáticos. La mazdutida se está investigando actualmente en los ensayos de fase 3 DREAM y GLORY para el tratamiento de la obesidad (NCT05607680) y la diabetes tipo 2 (NCT05606913).Como nota general, es esencial que las cohortes de ensayos clínicos se recluten de diversos orígenes étnicos. Existen diferencias bien establecidas en la prevalencia de diabetes, la incidencia de complicaciones metabólicas y las tasas de mortalidad entre los diferentes grupos étnicos. Las personas negras e hispanas presentan una mayor carga de diabetes tipo 2 y mayores tasas de complicaciones diabéticas, como enfermedades cardiovasculares. De hecho, las tasas de mortalidad por enfermedades cardiovasculares se encuentran entre las más altas en las personas negras con diabetes tipo 2. Por lo tanto, es esencial comprender mejor en qué se traducen las susceptibilidades diferenciales a las enfermedades, en particular con respecto a las diferentes respuestas metabólicas a los péptidos coagonistas en poblaciones de diferentes orígenes étnicos.
Cotadutida
La cotadutida (también denominada MEDI0382) es un péptido sintético basado en la OXM humana, que emplea una cadena lateral de ácido palmítico que permite la unión de la albúmina y, por lo tanto, la prolongación de su duración de acción. El péptido es notablemente más potente en GLP-1R, en relación con GCGR, con una proporción de ∼5:1. De manera similar a otros coagonistas de GLP-1R/GCGR, la cotadutida muestra una eficacia metabólica robusta en roedores y primates no humanos cynomolgus sanos, con superioridad en la pérdida de peso y el control de la glucosa, en comparación con el agonista selectivo de GLP-1R liraglutida. Esta mayor eficacia en la reducción del peso corporal se atribuye nuevamente a un aumento en el gasto de energía impulsado por el agonista de GCGR, asociado con una inducción de saciedad mediada por el agonista de GLP-1R. Más recientemente, se demostró que la cotadutida alivia la esteatosis hepática, reduce la fibrosis hepática y mejora la función mitocondrial en modelos murinos de enfermedad hepática esteatósica asociada a disfunción metabólica (MASLD), de manera mucho más efectiva que la liraglutida. Es de destacar que, para aumentar la conciencia sobre la enfermedad y prevenir el estigma, la terminología y los criterios de diagnóstico de la enfermedad del hígado graso no alcohólico (NAFLD) y la esteatohepatitis no alcohólica (NASH) fueron recientemente reemplazados por la nomenclatura MASLD y esteatohepatitis asociada a disfunción metabólica (MASH), respectivamente.] En conjunto, la cotadutida y otros coagonistas similares están firmemente posicionados como prospectos viables para el tratamiento de MASLD y MASH. Además, sus beneficios metabólicos indican que el agonismo mejorado del glucagón puede promover la deslipidación hepática, lo que debería tener grandes ventajas medicinales para el tratamiento de la obesidad, independientemente de la magnitud de la pérdida de peso. En estudios clínicos, la cotadutida fue de hecho bien tolerada, y en ensayos clínicos de fase 2, el péptido mostró una eficacia impresionante en la pérdida de peso, capacidades superiores para reducir los lípidos hepáticos y supresión del apetito, junto con una reducción de la presión arterial y los niveles de HbA1c en individuos con obesidad y diabetes tipo 2. Después de 32 días de tratamiento diario, la cotadutida mejoró aún más el control de la glucosa posprandial y potenció la pérdida de peso en sujetos con diabetes tipo 2 y enfermedad renal crónica, que fue paralela a una notable reducción del 51% en la relación albúmina-creatinina urinaria. Actualmente, la cotadutida se está evaluando en ensayos clínicos de fase 2b en sujetos con MASLD (PROXYMO-ADV, NCT05364931), con varios estudios de fase 2b adicionales completados en sujetos con (1) enfermedad renal crónica con diabetes tipo 2 (NCT04515849), (2) obesidad y MASLD/MASH (NCT04019561), o (3) obesidad con diabetes tipo 2 (NCT03555994), con resultados pendientes de publicación (Figura 3).
Otros coagonistas de GLP-1R/GCGR
NN9277/NN6177 (también denominado NN-117/NNC9204-1177) fue desarrollado por Novo Nordisk para el tratamiento de la obesidad y la diabetes de tipo 2. NN9277 completó tres ensayos clínicos de fase 1 en sujetos sanos, además de sujetos con sobrepeso y obesos, utilizando dosis múltiples que oscilaban entre 1 y 6 mg (NCT04059367, NCT03308721 y NCT02941042). En sujetos obesos, NN9277 mostró una eficacia impresionante en la pérdida de peso, con una reducción del peso corporal ajustada al placebo de aproximadamente el 12,6 % después de 12 semanas de tratamiento. Sin embargo, su desarrollo clínico finalmente se interrumpió debido a efectos adversos, en particular aumento de la frecuencia cardíaca y alteración de la tolerancia a la glucosa.ALT-801, anteriormente conocido como SP-1373, es un potente coagonista equilibrado con OW para GLP-1R y GCGR. El péptido se derivó químicamente con un glicolípido mediante el uso de tecnología de conjugación surfactante-péptido para retrasar la absorción y extender aún más su vida media. Este coagonista fue diseñado inicialmente para el tratamiento de MASLD y obesidad, lo que está respaldado por la observación de que después de 12 semanas de administración de péptidos en un modelo de ratón de obesidad y MASLD, fue evidente una reducción del peso corporal de ∼25%, concomitante con una mejora de la esteatosis hepática, la inflamación y la fibrosis.= Al igual que la cotadutida y la familia más amplia de agonistas multirreceptores basados en glucagón, estos resultados alientan una vía prometedora para ALT-801 en el tratamiento de MASLD asociada a la obesidad. De hecho, ALT-801, ahora denominado pemvidutida, se está examinando en un entorno clínico, ya que cinco ensayos están en curso o se han completado, todos en el contexto de la obesidad, la diabetes de tipo 2 o MASLD. En un estudio de fase 1 compuesto por 100 sujetos con sobrepeso y obesidad, 12 semanas de tratamiento con pemvidutida (1,8 mg) dieron como resultado una pérdida de peso corregida por placebo de aproximadamente el 10,3 % (NCT04561245). Actualmente, la pemvidutida también se está evaluando para el tratamiento de la obesidad en un estudio de fase 2 de dosis múltiples ascendentes de 48 semanas, compuesto por 320 sujetos obesos (ensayo de obesidad MOMENTUM) (NCT05295875).Otro coagonista de GLP-1R/GCGR, JNJ-64565111 (también denominado HM-12525A, efinopegdutida o MK-6024), se evaluó en varios estudios clínicos de fase 2 para el tratamiento de la obesidad y la diabetes de tipo 2 (NCT03586830). En sujetos con obesidad sin diabetes tipo 2, 26 semanas de tratamiento con JNJ-64565111, a la dosis más alta probada de 10 mg, dieron como resultado una pérdida de peso corregida por placebo de ∼10%, en relación con −5,8% en los controles tratados con liraglutida. En sujetos con obesidad y diabetes tipo 2, 12 semanas de tratamiento con JNJ-64565111 redujeron el peso corporal corregido por placebo en ∼7,2%, sin embargo sin efectos notables en los niveles de HbA1c o los niveles de glucosa plasmática en ayunas, aunque con una reducción apreciable en los niveles de insulina en ayunas. En ambos estudios, el tratamiento con JNJ-64565111 dio como resultado una mayor aparición de efectos adversos, especialmente náuseas y vómitos. La frecuencia de estos efectos secundarios ocurrió en el 84% y el 67% de los sujetos que recibieron JNJ-64565111 y en el 71% y el 48% de los sujetos recibieron liraglutida. Actualmente, se está evaluando JNJ-64565111 en comparación con semaglutida en un estudio de fase 2 en sujetos con MASLD (NCT04944992). Actualmente, se están examinando dos coagonistas GLP-1R/GCGR adicionales en ensayos clínicos; sin embargo, sus resultados metabólicos esperan publicación. En particular, BI-456906 (también denominado survodutida) es un péptido acilado graso que se está evaluando en ensayos de fase 2 para el tratamiento de la obesidad (NCT04667377) y la diabetes tipo 2 (NCT04153929). Además, el péptido coagonista OPK-88003 se está explorando en un estudio de fase 2 en sujetos con diabetes tipo 2 (NCT03406377).
En resumen, una gran cantidad de estudios preclínicos y clínicos (Figuras 2 y 3) se centran activamente en los coagonistas GLP-1R/GCGR, y la mayoría de los péptidos muestran un impacto impresionante en la reducción del peso corporal, la mejora del control glucémico y el mantenimiento o la restauración de la homeostasis metabólica. Será interesante seguir cómo estos coagonistas GLP-1R/GCGR particulares competirán en el campo con otras combinaciones de coagonistas basados en incretinas, además de la superioridad de los péptidos triagonistas de rápida inminencia en el futuro cercano. Otras preguntas se centran en si la combinación de tres agonistas de receptores objetivo resultará ventajosa con respecto a las mejoras metabólicas en la función cardiorrenal o hepática. Además, la pregunta es ¿cuál será la proporción más adecuada de afinidades relativas de los receptores para los péptidos coagonistas o triagonistas, que se traducirá en efectos adversos mínimamente asociados?
Coagonistas de GLP-1R/GIPRLa justificación para combinar el agonismo de GLP-1R y GIPR
Basados en la persistente observación de que los ratones deficientes en Gipr de la línea germinal están protegidos de la obesidad inducida por la dieta, combinado con los hallazgos que muestran que la acción insulinotrópica de GIP se ve ampliamente atenuada en sujetos con diabetes tipo 2, existe un debate persistente sobre si el receptor debe activarse o inhibirse para alcanzar un mérito metabólico óptimo. Por ejemplo, se ha demostrado que la inhibición farmacológica o genética del GIPR alivia la acumulación intramuscular de lípidos en ratones viejos. Además, mientras que algunas mutaciones de GIPR están asociadas con un índice de masa corporal más bajo en humanos, ciertos antagonistas de GIPR reducen el peso corporal y la ingesta calórica en ratones obesos inducidos por la dieta y primates no humanos, particularmente cuando se administran junto con el agonismo de GLP-1R. Sin embargo, mientras que también se observa protección contra la obesidad en ratones deficientes en Glp1r, la casi normalización de la hiperglucemia restaura el efecto insulinotrópico de GIP en sujetos con diabetes tipo 2. Por el contrario, el tratamiento con agonistas de GIPR es igualmente eficaz para promover la pérdida de peso en ratones obesos y, además, la pérdida de peso impulsada por GIP también se mejora sinérgicamente mediante el agonismo adjunto de GLP-1R. Es importante destacar que, en ratones con pérdida de Gipr en el SNC en general, la inhibición impulsada por GIP en la ingesta de alimentos disminuye por completo, mientras que la pérdida de peso inducida por GIP se restaura parcialmente. Esto indica que GIP impulsa la pérdida de peso a través de la inhibición central de la ingesta de alimentos y a través de mecanismos periféricos no relacionados con el SNC no relacionados con la ingesta de alimentos.
Evidencia adicional de los beneficios periféricos de la activación de GIPR proviene de observaciones recientes que muestran que los efectos antiinflamatorios de la activación de GLP-1R están mediados exclusivamente a través de las acciones centrales de GLP-1R. Sin embargo, el tratamiento de ratones deficientes en Glp1r específico del cerebro con un coagonista GLP-1R/GIPR conserva la acción antiinflamatoria, lo que sugiere que la actividad periférica de GIPR en el tejido adiposo y las células inmunes, es decir, los macrófagos, puede provocar efectos sistémicos positivos A la luz de esto, postularíamos que la activación periférica de GIPR en los adipocitos blancos tiene la capacidad de modular el equilibrio energético y la pérdida de peso. Para corroborar estas afirmaciones, se justifica un mayor trabajo con un enfoque en el tejido adiposo en el futuro para definir mejor las contribuciones que la activación periférica de GIPR, específicamente en el adipocito blanco, provoca hacia los beneficios metabólicos generales de estos agonistas. La razón para utilizar el agonismo de GIPR en un enlace unimolecular con GLP-1 fue, por lo tanto, doble. En primer lugar, la combinación del GLP-1R y el agonismo de GIPR podría mejorar aún más el metabolismo de la glucosa a través de una acción insulinotrópica y glucagonostática aditiva en el páncreas y potencialmente incluso restaurar la sensibilidad deteriorada al GIPR característica de la diabetes tipo 2. En segundo lugar, la combinación física de GLP-1 y GIP podría actuar en sinergia para superar al monoagonismo basado en GLP-1, produciendo así una mayor pérdida de peso con una mayor reducción en la ingesta de alimentos. Si bien la cuestión del valor terapéutico del GIP se ha visto obstaculizada por el debate en curso sobre si el GIPR debe activarse o inhibirse, ningún antagonista de GIPR ha recibido aún la aprobación regulatoria, y el éxito del coagonismo GLP-1R/GIPR, como se analiza a continuación, ha rehabilitado la opinión de que el agonismo de GIPR es un componente exitoso de las terapias basadas en incretinas.
Existen dos péptidos coagonistas unimoleculares de GLP-1R/GIPR que se han caracterizado preclínica y clínicamente por mostrar un potencial mejorado de reducción de glucosa, pérdida de peso superior y supresión del apetito cuando se evalúan con agonistas de GLP-1R comparables. Son NN0090-2746 (también conocido como NN9709, MAR709, RG7697 o RO6811135), y tirzepatida (inicialmente denominada LY3298176)84,154,155,156 . Los estudios preclínicos se centraron en si la adición de la activación de GIPR mejora o proporciona beneficios metabólicos únicos a través de mecanismos distintos del monoagonismo de GLP-1R.
Descubrimiento del primer coagonista de GLP-1R/GIPR
A lo largo de una década, se han producido importantes avances en el descubrimiento de coagonistas de GLP-1R/GIPR, que se basaron en la base de los agonistas de GLP-1R. En 2013, adelantándose a la curva del descubrimiento de fármacos multirreceptores para el tratamiento de la obesidad, se generó el primer coagonista de GLP-1R/GIPR, como un único péptido con un coagonismo potente y equilibrado en GLP-1R y GIPR Este péptido se construyó sobre una secuencia de glucagón que se modificó químicamente a un péptido con un agonismo comparable en GLP-1R y GIPR, pero sin actividad de GCGR. La primera generación de péptidos coagonistas de GLP-1R/GIPR se pegiló para respaldar la dosificación en OW. Este coagonista mostró una eficacia superior con efectos antihiperglucémicos e insulinotrópicos en ratones diabéticos db/db, ratas obesas diabéticas Zucker y primates no humanos cynomolgus. En ratones obesos inducidos por la dieta, el coagonista redujo de manera impresionante el peso corporal en aproximadamente un 26,9 %, en comparación con el 15,6 % después del tratamiento con liraglutida. El péptido redujo aún más la ingesta de alimentos, disminuyó la masa grasa y mejoró la esteatosis hepática en relación con la dosis equimolar con cualquiera de las incretinas solas (Figura 2). Esto destacó por primera vez que el coagonismo suplementario de GLP-1R/GIPR podría superar en gran medida al monoagonismo de GLP-1R para lograr una mayor pérdida de peso y generar mejoras adicionales en la homeostasis de la glucosa. Estudios posteriores implicaron la generación de una versión acilada grasa del péptido con una actividad potente equilibrada selectiva para GLP-1R y GIPR.157,158 Este péptido unimolecular, denominado MAR709, albergaba varias sustituciones de aminoácidos, una acilación en una lisina C-terminal, con un ácido palmítico C16 saturado. El péptido era resistente a la proteólisis de DPP-4 y mostraba un perfil farmacocinético adecuado para el uso clínico diario. Como testimonio farmacológico de la contribución vital del agonismo de GIPR en la acción metabólica del coagonista, el tratamiento de ratones obesos inducidos por la dieta con MAR709 produjo una mayor pérdida de peso y una mayor supresión en la ingesta de alimentos en relación con el tratamiento con la estructura principal de GLP-1 farmacocinéticamente coincidente; esta superioridad desapareció en ratones que presentaban una deleción de Gipr en el SNC, o más específicamente en neuronas GABAérgicas. Estos estudios identificaron esencialmente el sistema GIP cerebral como un nuevo regulador del metabolismo energético. En conjunto, estos estudios preclínicos pioneros establecieron el coagonismo GLP-1R/GIPR como una estrategia farmacológica alentadora para promover la pérdida de peso más allá de lo que se puede lograr con el agonismo de GLP-1R solo, sellando así el destino de las terapias multirreceptoras basadas en el intestino en un camino fructífero para futuros descubrimientos de poliagonistas. MAR709 completó con éxito los ensayos clínicos de fase 1 con buena tolerabilidad, concomitantemente con una reducción significativa en el peso corporal y los niveles de HbA1c en sujetos con diabetes tipo 2. Sin embargo, después de completar los ensayos de fase 2b, en los que una dosis única probada mostró solo una superioridad moderada en el peso corporal y el control de la glucosa sobre la liraglutida después de 12 semanas de tratamiento en sujetos con diabetes tipo 2, el desarrollo clínico de MAR709 se interrumpió a favor de continuar con el avance clínico del agonista de GLP-1R semaglutida.
Generación del segundo coagonista de GLP-1R/GIPR: tirzepatida
El segundo coagonista importante de GLP-1R/GIPR generado fue LY3298176, también denominado tirzepatida. Este péptido refleja la progresión en la optimización química de los agonistas de GLP-1R con la aplicación de un diácido graso acilado. Esta modificación había impulsado a la semaglutida a ser un agonista de GLP-1R de acción mucho más prolongada y de mayor potencia y eficacia que la liraglutida. De manera análoga, la tirzepatida representa una evolución similar en la estructura del coagonista de MAR709 al emplear un diácido graso análogo a la semaglutida en la misma ubicación en un péptido basado en GIP que se ha modificado para incluir la actividad de GLP-1. La vida media clínica de la tirzepatida es de aproximadamente 5 días (∼116,7 h), lo que permite la dosificación en OW. Una diferencia crítica entre tirzepatida y MAR709 es que MAR709 muestra una actividad «equilibrada» tanto en GLP-1R como en GIPR, mientras que tirzepatida favorece a GIPR humano sobre GLP-1R en una proporción de 5:1. En particular, MAR709 y tirzepatida presentan importantes diferencias específicas entre especies, ya que ambas moléculas son agonistas completamente activos en el receptor GIP humano; sin embargo, solo MAR709 es completamente activo en el receptor GIP de ratón. Como resultado, mientras que MAR709 requiere la señalización funcional de GIPR en el SNC para superar el agonismo de GLP-1R para reducir aún más el peso corporal y la ingesta de alimentos, la tirzepatida no reduce el peso corporal en ratones deficientes en Glp1r. Además, mientras que la tirzepatida promueve la secreción de insulina en islotes murinos exclusivamente a través del GLP-1R, el péptido estimula la secreción de insulina en islotes humanos predominantemente a través del GIPR. Sin embargo, en consonancia con la liberación de su potencial activador completo en el GIPR humano,[ estudios in vitro en células HEK293 humanas revelaron que la tirzepatida activa tanto la señalización de GIPR como la de GLP-1R, y en células β pancreáticas humanas aisladas, la secreción de insulina inducida por tirzepatida fue mayor, en relación con el tratamiento con GIP o GLP-1 solo. Los estudios in vivo demostraron que en ratones obesos inducidos por la dieta, la tirzepatida mejora la secreción de insulina dependiente de la glucosa, mejora la tolerancia a la glucosa, estimula la supresión del apetito y promueve una notable pérdida de peso, esto último como resultado del aumento de la oxidación de lípidos en los tejidos (Figura 2). Más específicamente, se informó que el tratamiento con tirzepatida impulsa un aumento específico del tejido en la eliminación de glucosa, preferentemente en el tejido adiposo blanco epididimario, el tejido adiposo marrón (BAT) y el músculo esquelético,un efecto asociado con la regulación positiva transcripcional en genes asociados con el catabolismo de aminoácidos de cadena ramificada en el BAT. Además, se demostró que el péptido induce un cambio en la ingesta de macronutrientes al reducir selectivamente la preferencia por una dieta rica en grasas y dulce a una preferencia por una dieta baja en grasas. En una nota similar, estudios en musarañas almizcleras y ratones revelaron que la administración de monoagonistas GIPR atenuó el efecto emético del agonismo de GLP-1R, una propiedad altamente deseable que puede contribuir a una mayor tolerabilidad de los medicamentos basados en GIP en relación con las monoterapias basadas en GLP-1 en dosis más altas.
Hallazgos clínicos de la tirzepatida
La FDA aprobó la tirzepatida para el tratamiento de la diabetes de tipo 2 en mayo de 2022 y, posteriormente, para el tratamiento de la obesidad en noviembre de 2023 (Figura 3). El péptido se ha evaluado ampliamente en numerosos ensayos clínicos, muchos de los cuales aún están en curso . Los ensayos multicéntricos SURPASS 1-6 evaluaron la eficacia de la tirzepatida (5, 10 o 15 mg por semana) para tratar la diabetes de tipo 2 en sujetos hispanos y blancos no hispanos con obesidad. SURPASS-1, por ejemplo, evaluó la eficacia de la tirzepatida en relación con el placebo en sujetos con obesidad y diabetes de tipo 2, que fueron reclutados en 52 hospitales de la India, Japón, México y los EE. UU. Después de 40 semanas de tratamiento, aproximadamente el 92 % de los sujetos que recibieron tirzepatida alcanzaron una HbA1c de <7,0 % en relación con el 19 % que recibió placebo, y el 52 % frente al 1 % de los sujetos alcanzaron una HbA1c de <5,7 %. Se observaron beneficios glucémicos similares en ensayos SURPASS posteriores, en los que la tirzepatida demostró ser superior en la mejora del control glucémico en relación con el tratamiento con semaglutida (1 mg), insulina degludec, insulina glargina e insulina lispro, de manera importante, con una eficacia preservada y sin comprometer la seguridad en sujetos con riesgo de enfermedades cardiovasculares. El ensayo SURPASS J-mono evaluó los efectos glucémicos de la tirzepatida en sujetos japoneses con diabetes tipo 2, demostrando que después de 52 semanas de tratamiento, los niveles de HbA1c se redujeron en un −2,8 %, en relación con el −1,3 % en los sujetos que recibieron dulaglutida. Se informaron mejoras impresionantes similares en la glucemia en el ensayo SURPASS J-combo, en el que se administró tirzepatida durante 52 semanas como terapia adicional a sulfonilureas, biguanidas, inhibidores de la α-glucosidasa, tiazolidinedionas, glinidas o inhibidores de la proteína transportadora de sodio-glucosa 2 (SGLT2) en sujetos japoneses con diabetes tipo 2 mal controlada. De manera similar, el ensayo SURPASS AP-combo examinó los efectos de la tirzepatida en comparación con la insulina glargina en sujetos con diabetes tipo 2 originarios de China, Corea del Sur, Australia e India. En conjunto, aún se están realizando varios ensayos SURPASS que probablemente logren resultados metabólicos positivos similares, incluidos (1) el ensayo SURPASS-CVOT en sujetos con diabetes tipo 2 que tienen antecedentes de enfermedad cardiovascular, (2) el estudio SURPASS-EARLY en sujetos con diabetes tipo 2 que fueron diagnosticados no más de 4 años antes del reclutamiento, (3) el ensayo SURPASS-SWITCH, que evalúa los efectos glucémicos de sujetos que cambiaron de dulaglutida a tirzepatida, y (4) el estudio SURPASS-PEDS, que examina los efectos metabólicos del tratamiento con tirzepatida en niños con diabetes tipo 2.Para evaluar la eficacia del tratamiento con tirzepatida principalmente en el contexto de la obesidad, se lanzaron los ensayos multicéntricos SURMOUNT . En el ensayo SURMOUNT-1 para sujetos obesos sin diabetes tipo 2, 72 semanas de tratamiento con tirzepatida lograron una pérdida de peso de hasta un −20,9 %, en relación con los controles con placebo. De manera similar, en un estudio con una duración de tratamiento comparable, aunque en sujetos con obesidad y diabetes tipo 2, la tirzepatida redujo el peso corporal en un −14,7 %, en relación con un −3,2 % en los controles con placebo. A pesar de las ligeras diferencias basadas en la cohorte de sujetos y la duración del tratamiento, esta magnitud de pérdida de peso fue en gran medida consistente con los ensayos SURPASS. En el ensayo SURMOUNT-3, se evaluaron los efectos de la tirzepatida en sujetos obesos sin diabetes tipo 2 que se sometieron a modificaciones intensivas del estilo de vida; El tratamiento terapéutico complementario con tirzepatida durante 72 semanas redujo el peso corporal en un −18,4%, en relación con un aumento de peso del 2,5% evidente en los controles con placebo. Los ensayos SURMOUNT en curso incluyen el SURMOUNT-MMO, en el que se controlará a los sujetos durante 5 años para detectar la aparición de eventos adversos cardiovasculares, además del ensayo SURMOUNT-OSA, en el que se evaluarán los efectos de la tirzepatida en la apnea obstructiva del sueño. Por último, ya estén en curso o en preparación los ensayos SUMMIT , que tienen como objetivo evaluar la acción de la tirzepatida para el tratamiento de la obesidad y sus enfermedades asociadas. Por ejemplo, el ensayo SUMMIT-HFpEF (insuficiencia cardíaca con fracción de eyección preservada) examinará a sujetos obesos con insuficiencia cardíaca, así como el estudio TREASURE-CKD que abordará la obesidad en el contexto de la enfermedad renal crónica.
El interés científico mundial en la tirzepatida está aumentando a un ritmo exponencial y, como tal, actualmente se están llevando a cabo otros ensayos clínicos que van más allá del alcance de esta revisión. En conjunto, los ensayos SURPASS y SURMOUNT han verificado que la seguridad de la tirzepatida es consistente con los agonistas de GLP-1R, junto con su eficacia mejorada en dosis máximas de 5, 10 y 15 mg . Otros estudios destacables con respecto a la tirzepatida incluyen ensayos de fase 1 que utilizan imágenes por resonancia magnética para evaluar las regiones centrales del cerebro que regulan la ingesta de alimentos y el gasto de energía junto con un estudio para examinar el vaciamiento gástrico en sujetos obesos o con diabetes tipo 2 . Recientemente, también se ha demostrado que el tratamiento con tirzepatida mejora la sensibilidad a la insulina, mejora la función de las células β pancreáticas y retarda las variaciones de glucosa durante las pruebas de tolerancia a las comidas en sujetos con diabetes tipo 2. Cabe destacar que la eficacia de la tirzepatida y la semaglutida para reducir el peso corporal se comparó recientemente en un estudio de metanálisis compuesto por datos de más de 41 000 personas. Después de 1 año de tratamiento, se logró una pérdida de peso de ≥10% en el 62,1% de los sujetos que recibieron tirzepatida y en el 38,0% de los sujetos que recibieron semaglutida. La evidencia preliminar sugirió además que el tratamiento con tirzepatida condujo a una pérdida de peso adicional de -4,3% después de 6 meses y -7,2% después de 12 meses, sin diferencias en la aparición de efectos adversos gastrointestinales.En conjunto, es evidente que los coagonistas de GLP-1R/GIPR han establecido firmemente un punto de referencia sin precedentes para los medicamentos destinados a tratar la diabetes de tipo 2 y la obesidad.La experiencia clínica con tirzepatida ha captado una enorme atención y ha alimentado mucho interés en el desarrollo de otras combinaciones de péptidos activadores de múltiples receptores. Aún quedan varias preguntas que se centran en el mecanismo definitivo por el cual los coagonistas de GLP-1R/GIPR logran sus notables resultados metabólicos. Se ha caracterizado preclínicamente que el agonismo de GIPR amplifica el rendimiento glucémico y de reducción de peso del coagonismo GLP-1R/GIPR, en comparación con el agonismo selectivo de GLP-1R, y, como tal, debe ser considerado un componente esencial que ayuda a lograr los beneficios metabólicos óptimos del coagonista. De hecho, con algunos ensayos clínicos programados para comparar GIP con semaglutida para la diabetes tipo 2 y la tolerancia a la glucosa en individuos con función de GIPR, GLP-1R y GLP-2R genéticamente alterada, es muy probable que en el futuro, los estudios clínicos que utilicen agonistas monorreceptores de GIPR informen beneficios metabólicos significativos en relación con la diabetes tipo 2 y la obesidad. También es igualmente plausible, aunque aún debe validarse clínicamente, que los agonistas de GIPR resulten exitosos en terapias combinadas con otros medicamentos no basados en incretinas. Sin embargo, la cuestión específica de si el agonismo requiere la integración molecular en una sola molécula o puede utilizarse en una proporción ajustable aún no se ha abordado por completo. Otros coagonistas unimoleculares en desarrollo clínico incluyen una formulación oral de un péptido coagonista GLP-1R/GIPR, patrocinada por Novo Nordisk, que se encuentra en ensayos clínicos de fase 1 para sujetos con diabetes tipo 1. De manera similar, la combinación de dosis fija (FDC) Sema-OW GIP, es una combinación inyectable subcutánea de semaglutida con un nuevo agonista GIPR, denominado NNC0480-0389. Esta combinación de fármacos se está estudiando actualmente en sujetos sanos, además de en individuos obesos y con diabetes tipo 2 (NCT04259801), y se prevé que entre en ensayos de fase 2 en un futuro próximo. En conjunto, ahora es evidente que al asociar GLP-1R y GIPR para generar coagonistas basados en incretinas se logran resultados metabólicos espectaculares con una dosis terapéutica mejorada. La progresión en el rendimiento de estos coagonistas GLP-1R/GIPR sigue la tendencia química desarrollada primero en el agonismo GLP-1R con liraglutida y semaglutida para ahora lograr una secuela metabólica mucho mayor.Tomados en conjunto, dos péptidos GLP-1R/GIPR brillaron en el centro de atención de la investigación clínica de los coagonistas unimoleculares sofisticados y, como tal, se debe dar gran crédito a estos desarrollos iniciales que crearon una base sólida para los muy esperados péptidos triagonistas multirreceptores que vendrían después. Finalmente, si bien los mecanismos precisos que subyacen a los beneficios coordinados del coagonismo GLP-1R/GIPR aún deben desentrañarse por completo, es concebible que el tejido adiposo como un objetivo periférico pueda desempeñar un papel significativo en la regulación del equilibrio energético, dado el grado considerable de reducción de peso y pérdida de masa grasa. Como tal, los estudios preclínicos futuros sobre la base mecanística de la acción de los péptidos coagonistas deberían resultar esclarecedores.
Triagonistas GLP-1R/GIPR/GCGR
Basándose en el éxito preclínico de los coagonistas GLP-1R/GCGR y los coagonistas GLP-1R/GIPR, era naturalmente intuitivo suponer que un único péptido que muestra una actividad equilibrada en los tres receptores diana mejoraría aún más el control glucémico y aceleraría la pérdida de peso, con la esperanza de superar lo que ya se había logrado con la cirugía bariátrica. El desafío químico, sin embargo, era satisfacer los requisitos estructurales para el agonismo en tres receptores relacionados pero diferentes, donde las hormonas nativas GLP-1, GIP y glucagón son altamente específicas en sus interacciones. Cabe destacar que la farmacología de la primera generación de dichos triagonistas unimoleculares ya demostró ser superior a cualquier péptido único o coagonista de primera clase en ese momento (Figura 2).
Hallazgos preclínicos que dieron inicio al triagonismo unimolecular.Estudios preclínicos informativos que comenzaron mucho antes de que se lograra la pérdida de peso en humanos, que ahora supera el 20 % con los mejores coagonistas de su clase, describieron la síntesis y caracterización de varios triagonistas unimoleculares GLP-1R/GIPR/GCGR. Estos hallazgos preclínicos prepararon el terreno para los ensayos clínicos en curso en obesidad y diabetes tipo 2 utilizando péptidos triagonistas. El primer triagonista unimolecular GLP-1R/GIPR/GCGR establecido preclínicamente fue MAR423. Este péptido fue protegido del reconocimiento de DPP-4 a través de un ácido aminoisobutúrico en la posición 2, mientras que la lisina en la posición 10 fue acilada grasa con un ácido palmítico a través de un enlazador de ácido γ-glutámico. Se introdujeron sustituciones de aminoácidos distintas en el centro del péptido para restaurar la actividad equilibrada del receptor de glucagón, mientras que el extremo C-terminal de la exendina-4 se unió para mostrar un agonismo completo equilibrado en los tres receptores. En ratones obesos inducidos por la dieta, MAR423 mostró impresionantes efectos de reducción del peso corporal dependientes de la dosis de 26,6% en 20 días, en comparación con 15,7% con el tratamiento con coagonistas GLP-1R/GIPR. El péptido redujo aún más la ingesta de alimentos y la masa grasa, mejoró el control glucémico, redujo la hipercolesterolemia y mejoró el metabolismo lipídico hepático, en relación con el agonismo de GLP-1R o el coagonismo equilibrado de GLP-1R/GIPR181 (Figura 2). Los ratones que carecían individualmente de cada uno de los tres receptores, o del antagonismo farmacológico de cada receptor, confirmaron la relevancia funcional de cada una de las tres entidades peptídicas, lo que fue más evidente con la inducción del gasto energético y la utilización de lípidos por parte del glucagón. MAR423 mejoró aún más la dislipidemia y mejoró la esteatosis hepática en ratones obesos, en particular incluso en dosis en las que el fármaco solo tuvo efectos marginales sobre el peso corporal y la saciedad.Tras la publicación del primer triagonista, surgió una serie de péptidos similares, pero con diferencias notables en la duración de la acción y la actividad en cada receptor diana. Tras 2 semanas de tratamiento, un triagonista de segunda generación refinado bioquímicamente mostró una impresionante pérdida de peso de >30% en roedores obesos inducidos por la dieta, con una pérdida de peso superior en relación con los mejores coagonistas de GLP-1R/GIPR. Este El triagonista GLP-1R/GIPR/GCGR, LY3437943, se basa en la secuencia GIP, en la que se introdujeron sustituciones de aminoácidos de forma gradual para lograr un triple agonismo. El péptido de 39 aminoácidos lleva aminoácidos no naturales en las posiciones 2, 13 y 20, que no solo protegen de la degradación mediada por DPP-4 sino que también mejoran la actividad en los receptores de GIP y glucagón. Un diácido graso C20 se ancló además en la lisina en la posición 17 para mejorar la biodisponibilidad a través de la unión de la albúmina. A diferencia del triagonista equilibrado MAR423, LY3437943 mostró una actividad equilibrada para GCGR y GLP-1R pero una potencia mejorada en el GIPR. En ratones obesos inducidos por la dieta, LY3437943 redujo de manera muy efectiva el peso corporal en aproximadamente un 45 %, que se mantuvo en gran medida en la termoneutralidad. La pérdida de peso estuvo acompañada además por una reducción en la masa grasa, una supresión transitoria en la ingesta de alimentos, junto con marcadas mejoras en el control glucémico. En particular, el péptido triagonista LY3437943 superó al coagonista GLP-1R/GIPR tirzepatide para lograr un mayor grado de pérdida de peso, una observación atribuida a un aumento en el gasto de energía, que notablemente representó aproximadamente entre el 30 % y el 35 % del peso perdido en ratones obesos inducidos por la dieta y disminuyó con la inhibición basada en anticuerpos del GCGR.
SAR441255 es un triagonista GLP-1R/GIPR/GCGR unimolecular balanceado sintéticamente, basado estructuralmente en la secuencia de exendina-4 con sustituciones seleccionadas y acilación de ácido palmítico. En primates no humanos cynomolgus delgados, la tomografía por emisión de positrones reveló una mayor ocupación del receptor para GLP-1R y GCGR. En ratones obesos inducidos por la dieta, se demostró que SAR441255 alivia la hiperglucemia y reduce el peso corporal en aproximadamente un 14,1 %; Además, cuando se comparó con un coagonista de GLP-1R/GCGR, este efecto se atribuyó principalmente a un aumento en el gasto de energía. También se observaron resultados metabólicos superiores similares en primates no humanos cynomolgus obesos y diabéticos.HM15211 (también denominado agonista triple LAP) es un péptido triagonista GLP-1R/GIPR/GCGR de acción prolongada basado en la secuencia de glucagón y conjugado con un fragmento cristalizable de glicosilato humano. HM15211 se examinó en modelos de primates no humanos cynomolgus y roedores de obesidad, diabetes tipo 2 y MASLD. Este triagonista superó a la liraglutida en términos de pérdida de peso y mejoró aún más la hiperglucemia, aumentó el gasto de energía, redujo la hipercolesterolemia y mejoró la esteatosis hepática y la fibrosis. Los últimos beneficios metabólicos se atribuyeron a la propiedades antiinflamatorias y su capacidad para reducir la producción del factor de crecimiento transformante β. En consonancia con estos hallazgos, una triple mezcla física separada de GLP-1R, GIPR y monoagonistas de GIPR también sirvió para amortiguar la acumulación de triglicéridos hepáticos en un modelo de ratón de MASLD y fibrosis. Por lo tanto, estos resultados resaltan el tremendo potencial de los péptidos triagonistas para tratar la MASLD asociada a la obesidad. Otros triagonistas menos caracterizados incluyen YAG/glucagón, [D-Ala2]GLP-1/glucagón, y [D-Ala2]GIP/Oxm.En conjunto, los estudios preclínicos identificaron que los péptidos triagonistas unimoleculares son muy superiores a los coagonistas y monoagonistas existentes en la regulación del peso corporal, la saciedad, el metabolismo lipídico hepático y el control glucémico (Figura 2). La contribución única de cada hormona permite resultados metabólicos sincronizados mejorados. La lógica mecanicista preclínica actual de cómo la actividad de cada receptor contribuye a la pérdida de peso podría ser la siguiente: (1) el agonismo de GLP-1R sirve principalmente para reducir la ingesta de alimentos y mejorar el control glucémico, (2) el agonismo de GCGR estimula el aumento del gasto energético, y (3) el agonismo de GIPR puede servir como un potenciador metabólico para potenciar los efectos de saciedad de GLP-1, mejorar la sensibilidad a la insulina y amortiguar la predisposición hiperglucémica del glucagón para permitir un agonismo más agresivo de GCGR. La biología celular básica de GIPR es probablemente la más compleja y desconcertante de los tres receptores. En esta etapa, no podemos excluir las acciones específicas de GIPR en la periferia, particularmente en el tejido adiposo, donde necesitamos obtener una mejor comprensión de lo que el receptor es capaz de lograr tras la activación en la grasa blanca.
Hallazgos clínicos en el desarrollo de péptidos triagonistas
En un entorno clínico, los péptidos triagonistas GLP-1R/GIPR/GCGR que progresaron hasta el desarrollo clínico incluyeron MAR423, LY3437943 (retatrutida), SAR441225, y HM15211 ( Figura 3). El primero en avanzar hasta el estudio clínico fue MAR423 (también conocido como NN9423), que, sin embargo, debido a su necesidad de administración una vez al día, se ha abandonado en favor de una versión oral.
Se ha demostrado que una dosis única de otro péptido triagonista, retatrutida, produce una pérdida de peso impresionante de hasta -3,52 kg con una dosis de 6 mg, que persiste hasta 6 semanas. Se logró una pérdida de peso comparable con tirzepatida después de cuatro dosis semanales. Curiosamente, los sujetos sanos mostraron una supresión transitoria del apetito, una reducción de los niveles endógenos de glucagón, un aumento de los niveles sistémicos de β-hidroxibutirato, junto con un perfil lipídico mejorado. En un estudio de prueba de concepto de fase 1 en sujetos con diabetes tipo 2, se exploró la eficacia de dosis ascendentes OW de retatrutida (0,5-12 mg), en comparación con dulaglutida (1,5 mg) o placebo (NCT04143802). Después de 12 semanas de tratamiento, retatrutida redujo los niveles absolutos de HbA1c en -1,90%, en comparación con -0,96% y -0,34% con dulaglutida o placebo, respectivamente. Retatrutida indujo además una pérdida de peso dependiente de la dosis de -8,65 kg desde el valor inicial con la dosis más alta194 . En estos estudios, retatrutida mostró un perfil de seguridad no abiertamente diferente al agonismo selectivo de GLP-1R, siendo algunas náuseas el efecto adverso notificado con mayor frecuencia. El triagonista redujo aún más la presión arterial sistólica y diastólica, con un aumento transitorio sutil de la frecuencia cardíaca; Este último es consistente con la terapéutica basada en GLP-1R. En conjunto, estos estudios clínicos iniciales indicaron que la retatrutida tiene un gran potencial para un rendimiento diferencial, en relación con la semaglutida y la tirzepatida, y el péptido actualmente constituye la vanguardia en la terapia de la obesidad basada en incretinas.Más recientemente, en un ensayo de fase 2, la retatrutida logró un récord sorprendente de ∼24,2% de pérdida de peso corregida con placebo en sujetos obesos sin diabetes tipo 2 después de 48 semanas de tratamiento. Es el punto de referencia actual para la mayor pérdida de peso jamás informada con esta clase de candidatos a fármacos contra la obesidad. En comparación, la pérdida de peso corregida con placebo inducida por tirzepatida o semaglutida en este punto temporal del tratamiento en una población de estudio comparable fue de ∼17% y ∼13%, respectivamente. En un estudio de fase 2 publicado consecutivamente compuesto por sujetos con obesidad y diabetes tipo 2, 36 semanas de tratamiento con retatrutida redujeron el peso corporal en aproximadamente un 16,9 %, en comparación con aproximadamente un 2 % o un 3 % con dulaglutida o placebo, respectivamente; además, el triagonista redujo los niveles de HbA1c en aproximadamente un 2 %, en comparación con aproximadamente un 1,4 % o un 0,01 % con dulaglutida o placebo, respectivamente, en el punto de tiempo de 24 semanas. Finalmente, en un subestudio de fase 2 en sujetos con MASLD, la retatrutida disminuyó significativamente la grasa hepática, mostrando una reducción sustancial de hasta un 86 % durante un período de 48 semanas, lo que sugirió que el péptido tiene el potencial de resolver la MASLD. En conjunto, estos estudios clínicos resaltan el poderoso impacto que tiene la retatrutida en el metabolismo de la glucosa y los lípidos, con un nivel sin precedentes de pérdida de peso que parece continuar al final del estudio.El tercer triagonista unimolecular de acción prolongada GLP-1R/GIPR/GCGR en desarrollo clínico es HM15211 (Figura 3). En un estudio de fase 1, se informó que la adición de actividad GIPR al coagonismo GLP-1R/GCGR potenciaba en gran medida la eficacia de reducción de peso y glucémica en sujetos con sobrepeso (NCT04521738). En un ensayo de fase 1 posterior, se utilizarán múltiples dosis ascendentes de HM15211 en 66 sujetos obesos con MASLD (NCT03744182). De manera similar, en un estudio de fase 2 en curso, se evaluarán 217 sujetos con MASH confirmado por biopsia (NCT04505436) . Este último ensayo clínico es una investigación de referencia importante, ya que HM15211 muestra específicamente mucho potencial en la enfermedad hepática relacionada con la obesidad, como un punto de distinción para el enfoque más habitual en el control de la glucosa y las capacidades de reducción del peso corporal.
Preguntas pendientes en torno al triagonismo unimolecular
Los objetivos medicinales de los triagonistas unimoleculares eran aprovechar tres mecanismos de señalización complementarios en la reducción de peso para lograr una eficacia sin precedentes, que pudiera rivalizar o incluso superar a la cirugía bariátrica. Sin embargo, una pregunta en particular es ¿cuántas de las tres actividades del receptor exhiben mecanismos independientes o superpuestos en la pérdida de peso?Inicialmente, la espectacular pérdida de peso lograda con el coagonismo GLP-1R/GIPR planteó la pregunta de cuál era la mejor manera de integrar el agonismo de GCGR. El impasse biológico de que el glucagón puede inducir hiperglucemia es un freno inmediato y obvio al grado de agonismo de GCGR. Además, el glucagón tiene efectos vasculares que deben manejarse con éxito o se arriesga la mejora total de los resultados cardiovasculares logrados con formas de terapia menos agresivas. Sin embargo, la mayor apreciación del papel que el glucagón tiene en el gasto energético ha colocado a esta hormona bajo una nueva luz y ha reavivado su consideración como un componente que podría proporcionar una pérdida de peso adicional cuando sea necesario o para sostener la pérdida de peso que de otro modo podría disminuir mediante alguna forma de suplementación. Un informe reciente también plantea potentes efectos renoprotectores que se ejercen a través del GCGR en el riñón. Los resultados clínicos con respecto a la tirzepatida parecen anclar la actividad de GIPR tanto como la actividad de GLP-1 en el tratamiento de la obesidad. Sin embargo, quedan dudas sobre si el GIPR es predominantemente un potenciador metabólico del GLP-1 o, con una eficacia sostenida, emerge cada vez más como un contribuyente primario para mantener la salud metabólica. La importancia del GIP en el triple agonismo puede ser doble, donde estos péptidos parecen tolerar la actividad del glucagón mucho más que el coagonismo GLP-1R/GCGR. El papel de la actividad de GIPR dentro del triagonista puede resultar más complejo y, aunque en esta etapa es especulativo, su función podría incluir la estimulación de vías cíclicas inútiles que desperdician energía. Sin duda, futuros estudios genéticos arrojarán luz sobre la función de GIP. Pero por ahora, la historia más amplia del desarrollo de fármacos contra la obesidad indica que debemos avanzar con la debida cautela, ya que ha habido otras formas de farmacología que han promovido la pérdida de peso de manera insegura. La tarea que tenemos por delante es utilizar estas herramientas farmacológicas y las observaciones mecanicistas preclínicas de la manera más inteligente e informada en el tratamiento de personas obesas y con diabetes tipo 2.
Efectos secundarios potenciales de los agonistas mono y multirreceptores
Un aspecto importante a considerar es que existe un amplio consenso en el campo terapéutico de que las intervenciones basadas en incretinas no pueden restablecer el “lipostat”, es decir, al suspender el tratamiento farmacológico, tanto en modelos de roedores como en estudios clínicos, se observa con frecuencia una recuperación del peso corporal, ya que el peso perdido finalmente vuelve a los niveles iniciales. En otras palabras, existe una alta tasa de “reincidencia” similar a la que se observa en los ensayos de intervención del estilo de vida. De hecho, la interrupción de semaglutida o tirzepatida conduce a un repunte significativo en la ganancia de peso corporal. Sin embargo, esto no debería ser una respuesta completamente inesperada, dado que se observan efectos similares después de la retirada del tratamiento de las enfermedades cardiometabólicas, es decir, para la hipertensión o la hipercolesterolemia. Si bien esta noción es generalmente cierta, hay evidencia reciente que sugiere que cierto grado de la pérdida de peso inicial puede conservarse después de un período de eliminación del fármaco. En el ensayo SURMOUNT-4, los sujetos obesos que fueron tratados con tirzepatida durante 36 semanas mostraron una reducción en el peso corporal de ∼21%. Los individuos que recibieron tirzepatida durante 52 semanas adicionales perdieron un 6% adicional, mientras que los sujetos que recibieron placebo durante el período de seguimiento de 52 semanas recuperaron el 14% de su peso corporal. A pesar de esta recuperación de peso, los sujetos a los que se les administró placebo aún mantuvieron una pérdida de peso de ∼10% en comparación con su peso corporal inicial, lo que es muy significativo. Sin embargo, en esta etapa, no se sabe si los pesos corporales volverán completamente a los niveles iniciales después de un período de seguimiento más prolongado. En conjunto, esto es una constatación seria de que las nuevas farmacoterapias para la pérdida de peso, a pesar de su eficacia sin precedentes, pueden no considerarse la cura definitiva para la obesidad. Sin embargo, en la actualidad, esto sugiere que para cosechar los beneficios completos de la pérdida de peso, puede ser necesaria la exposición al fármaco durante toda la vida. Sin embargo, esta perspectiva plantea el desafío de la farmacoterapia de próxima generación que, con suerte, ofrecerá una solución de tratamiento permanente para la obesidad, incluso después de la interrupción del tratamiento.Otra preocupación es que el grado de pérdida de peso inducido por el tratamiento farmacológico no se debe exclusivamente a una pérdida de masa grasa; más bien, existe una cantidad significativa de pérdida muscular que puede acompañar esto. Un debate en curso es si esta pérdida de masa magra es simplemente un reflejo de que una reducción en el peso corporal requiere menos masa muscular para proporcionar un soporte mecánico general o si la pérdida de masa magra plantea un problema de salud significativo para el individuo. Esto puede ser un problema considerable para las personas que entran y salen repetidamente del tratamiento farmacológico, lo que resulta en un fenómeno de «peso corporal yoyo», en el que cada ciclo se asocia con un aumento de la masa grasa, junto con una reducción de la masa muscular. Este aspecto es más preocupante en las poblaciones de pacientes mayores que sufren sarcopenia relacionada con la edad, incluso antes de la exposición a agonistas mono o multirreceptores. Sin embargo, estos problemas podrían atenuarse con una serie de contramedidas, como la inhibición del eje del receptor de activina tipo II o la activación de la vía del receptor de apelina. En la misma línea, sigue sin conocerse el rendimiento relativo de los agonistas mono o multirreceptores en subpoblaciones de diabetes tipo 2, por ejemplo, sujetos delgados con diabetes tipo 2. La diabetes tipo 2 delgada es una entidad clínica distinta y, dada la pérdida de peso de más del 20 % que se puede lograr en sujetos obesos con diabetes tipo 2, queda por determinar si dosis más bajas de tratamiento farmacológico pueden mantener el control glucémico y minimizar la pérdida de peso injustificada, ya que esto podría estar potencialmente asociado con una reducción de la masa magra en sujetos delgados con diabetes tipo 2.Por último, debemos mencionar los problemas logísticos y socioeconómicos asociados con el uso generalizado de estas nuevas intervenciones farmacológicas. En la actualidad, todavía prevalecen los problemas de suministro, ya que la demanda supera ampliamente la oferta. Sin lugar a dudas, el tiempo y un mercado más competitivo resolverán estos problemas. Sin embargo, lo que seguirá siendo un problema son los altos costos mensuales que las personas deben asumir, en vista del hecho de que la mayoría de los planes de seguro de salud no cubren los costos de los medicamentos contra la obesidad. La principal preocupación es que esto conduce a mayores disparidades en materia de salud, ya que los pacientes que más necesitan estas intervenciones se encuentran en la imposibilidad de pagarlas o de tener acceso a ellas.
Observaciones finales y perspectivas futuras
El progreso observado en la última década en el tratamiento de la obesidad y sus enfermedades relacionadas ha sido nada menos que asombroso. Los avances en el metabolismo durante el último siglo se aceleraron en las últimas décadas gracias a las biotecnologías colectivas para cumplir con la hipótesis de la incretina. El GLP-1 demostró ser sumamente eficaz en el tratamiento de la hiperglucemia asociada a la diabetes de tipo 2, sin el riesgo de hipoglucemia comúnmente asociado con la insulina y las sulfonilureas. A través de mejoras iterativas de última generación en sus propiedades farmacéuticas, surgieron agonistas peptídicos selectivos de GLP-1R altamente eficaces para el tratamiento de la diabetes de tipo 2. El enfoque terapéutico luego pasó a la obesidad, ya que las observaciones preclínicas y clínicas revelaron la reducción del peso corporal como un beneficio complementario en el tratamiento de la obesidad asociada a la diabetes de tipo 2. Las primeras formas de terapia demostraron ser comparablemente efectivas a las formas convencionales no peptídicas al proporcionar una reducción de un dígito medio en el peso corporal, con reducciones en los eventos cardiovasculares adversos. El paso fortuito hacia adelante se produjo cuando la semaglutida, un péptido diseñado para aumentar la comodidad del paciente a la hora de reducir la dosis inyectable de diaria a semanal, demostró ser doblemente eficaz para reducir el peso corporal a más del 10%, en comparación con una dosis diaria del péptido químicamente relacionado, la liraglutida. Al superar clínicamente la eficacia antiobesidad de la liraglutida, la semaglutida transformó la visión de lo que era farmacéuticamente posible y, por lo tanto, lanzó el tratamiento medicinal de la obesidad, a pesar de alcanzar solo una fracción de lo que se puede lograr mediante la cirugía bariátrica. Poco después, el rendimiento clínico de la semaglutida en el tratamiento de la diabetes de tipo 2 y/o la obesidad se vio sustancialmente eclipsado por la farmacología integrada de GIP y GLP-1 en la tirzepatida. El péptido sirvió para duplicar la convicción de la perspectiva de una eficacia para reducir el peso corporal comparable a la de la cirugía bariátrica, un objetivo que estaba al alcance, por primera vez, mediante el uso de la farmacología de múltiples mecanismos. Si bien los resultados clínicos validaron esta perspectiva, el breve intervalo de tiempo entre la semaglutida y la tirzepatida es una manifestación de que las semillas de la invención se habían plantado más de una década antes, con observaciones preclínicas que predijeron este resultado y un rendimiento aún mayor con más iteraciones en los mecanismos de acción. Con respecto al agonismo de GIPR, si bien aún no se ha informado un mecanismo definitivo sobre cómo la señalización de GIPR afecta el equilibrio energético, la activación de GIPR se ha asociado con una mejor salud del tejido adiposo. Considerando el papel destacado del tejido adiposo en la homeostasis energética, es plausible que el agonismo de GIPR en la grasa pueda aprovechar los efectos beneficiosos en la homeostasis metabólica.El avance más allá del agonismo selectivo de GLP-1 también estuvo arraigado en la controversia. Empezando por el agonismo del glucagón, que es contraintuitivo a la farmacología diabetogénica del glucagón, con décadas de trabajo en la literatura que persiguieron sin éxito su antagonismo para el tratamiento de la diabetes. La comprensión más profunda de que el glucagón, al igual que la insulina, tiene un alcance farmacológico más amplio que el mero manejo agudo de la glucosa, junto con la integración con GLP-1 y GIP, ha iluminado la posibilidad de utilizar la hormona con fines constructivos. No obstante, debe abordarse con gran cautela, en particular en relación con sus posibles efectos cardiovasculares, que son más difíciles de evaluar y potencialmente más difíciles de revertir que su impacto en el control de la glucemia. El éxito con GLP-1 y el éxito emergente similar con GIP no deberían disminuir nuestra preocupación en el manejo del glucagón, ya que las primeras son incretinas fisiológicas y más parecidas que la segunda hormona. La integración del agonismo del glucagón y del GIP en la farmacología del GLP-1 representa la forma más avanzada de terapia multimodal que está logrando un progreso extraordinario y actualmente está avanzando hacia la última etapa del desarrollo clínico. La Figura 3 destaca algunas de las terapias de próxima generación para el tratamiento de la obesidad y la diabetes tipo 2, con las contribuciones relativas del GLP-1, el glucagón y el GIP a las hormonas intestinales en cada péptido agonista y la notable pérdida de peso lograda con cada uno.
Las observaciones actuales de que el agonismo y el antagonismo del GIP pueden lograr resultados preclínicos similares en la reducción del peso corporal son desconcertantes. El papel fisiológico del GIP en otras funciones endocrinas, como el mantenimiento de la salud ósea, es de una importancia apreciable. Por ejemplo, el agonismo del GIPR dentro de los agonistas multirreceptores mejora el metabolismo óseo. Como tal, la perspectiva no deseada de que la inhibición de la actividad del GIP en una población de pacientes con diabetes tipo 2 con densidad mineral ósea reducida podría representar un riesgo adverso crónico que necesita ser abordado. Un estudio reciente que utilizó un anticuerpo bloqueador del GIPR conjugado con un potente agonista del receptor de GLP-1 en sujetos obesos ha reavivado el interés en dicha terapia multimodal. La eficacia sostenida de reducción del peso corporal en dosis mensuales que son más de 100 veces superiores a las que se emplean comúnmente con agonistas selectivos del GLP-1, sin efectos adversos gastrointestinales, es difícil de explicar. Sin duda, estudios futuros delinearán cómo es posible esta aparente discrepancia. En vista de esto, por el momento, parece mejor atenerse a la sabiduría de que “la prueba de una inteligencia de primer nivel es la capacidad de mantener dos ideas opuestas en la mente al mismo tiempo y aún así conservar la capacidad de funcionar”.En conclusión, la cirugía bariátrica sigue siendo el punto de referencia para la pérdida de peso, desencadenando una reducción de peso de aproximadamente el 25% al 30% en un período de 1 a 2 años.= Se ha informado que la semaglutida tiene capacidades de reducción de peso de aproximadamente la mitad de esta magnitud, y la tirzepatida redujo significativamente la diferencia relativa restante con la cirugía a la mitad. Los resultados preclínicos y los datos clínicos emergentes con agonistas triples indican más avances. No hay duda de que se han logrado avances importantes en el camino hacia el éxito de los agonistas multirreceptores como agentes terapéuticos para el tratamiento de la obesidad y la diabetes. Es un momento emocionante, ya que estos medicamentos se están trasladando a poblaciones más grandes más allá de los ensayos clínicos y, con suerte, resistirán la prueba del tiempo en la práctica de la vida real. La promesa de una terapia antiobesidad con este nivel de eficacia para reducir la carga de diabetes tipo 2 y las enfermedades cardiovasculares relacionadas, así como un posible impacto en la enfermedad renal crónica, el cáncer, la osteoartritis, el manejo del dolor y otras secuelas para las que el exceso de peso corporal es un factor de riesgo, está comenzando a ponerse a prueba en el mundo real. Las terapias con agonistas multirreceptores como Retatrutida ,se reconocen a partir de ahora como una vía esperanzadora que, sin duda, cambiará la trayectoria del incesante aumento de la obesidad mundial.

Figura 1. Vías de señalización y procesos metabólicos sobre los que actúan las hormonas endógenas GLP-1, GIP y glucagón en los tejidos dianaEfectos metabólicos específicos de los tejidos del péptido similar al glucagón-1 (GLP-1) (círculos azules), el péptido insulinotrópico dependiente de la glucosa (GIP) (círculos naranjas) y la hormona pancreática glucagón (GCG) (círculos rojos) de la incretina endógena secretada por el intestino. Se muestran las acciones principales de GLP-1, GIP y GCG en el tejido adiposo, el cerebro, el hígado, el páncreas, los huesos, el estómago y el corazón. Los cuadrados en negrita resaltan el cerebro y el tejido adiposo, dos de los sitios principales de la acción del agonista de GLP-1R, el coagonista de GLP-1R/GIPR y el coagonista de GLP-1R/GCGR en la regulación del peso corporal. Los círculos sólidos representan la acción directa de cada hormona, mientras que los círculos parciales resaltan su acción indirecta. Un signo rojo (−) destaca una función inhibidora de GLP-1, GIP y GCG, mientras que un signo verde (+) representa una función estimuladora de cada hormona. Cada proceso metabólico primario, vía de señalización y/o resultado metabólico que se ve afectado por GLP-1, GIP o GCG, ya sea de forma directa o indirecta, se resalta para cada tejido.
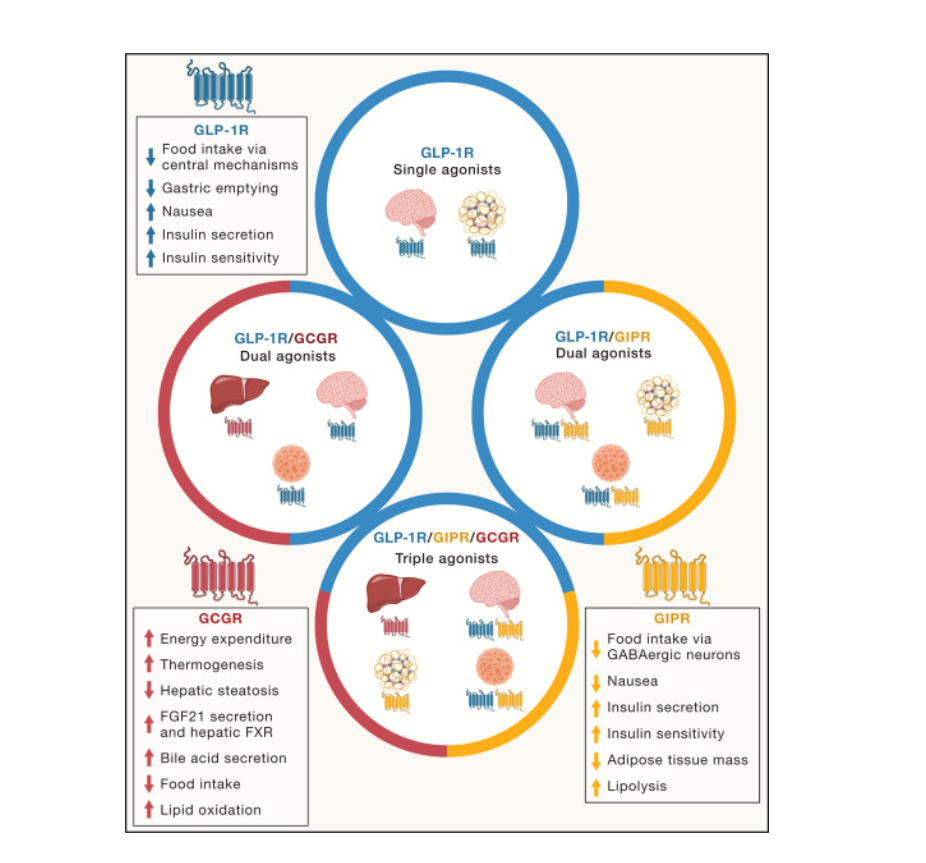
Figura 2. Principales sitios y acción específica de tejido de los monoagonistas de GLP-1R, coagonistas de GLP-1R/GCGR, coagonistas de GLP-1R/GIPR y triagonistas de GLP-1R/GIPR/GCGR determinados en estudios preclínicos
Los principales sitios de expresión y acción del receptor y los efectos farmacológicos que median la pérdida de peso corporal y las mejoras en el control glucémico de los agonistas de GLP-1R (texto azul y círculo), coagonistas de GLP-1R/GCGR (texto azul/rojo y círculo), coagonistas de GLP-1R/GIPR (texto azul/amarillo y círculo) y triagonistas de GLP-1R/GIPR/GCGR (texto azul/amarillo/rojo y círculo). Los estudios preclínicos han informado los efectos metabólicos, las vías de señalización y los posibles reguladores clave a los que se dirige cada agonista mono o multirreceptor en el cerebro, los islotes pancreáticos, el tejido adiposo, el hígado y el riñón.
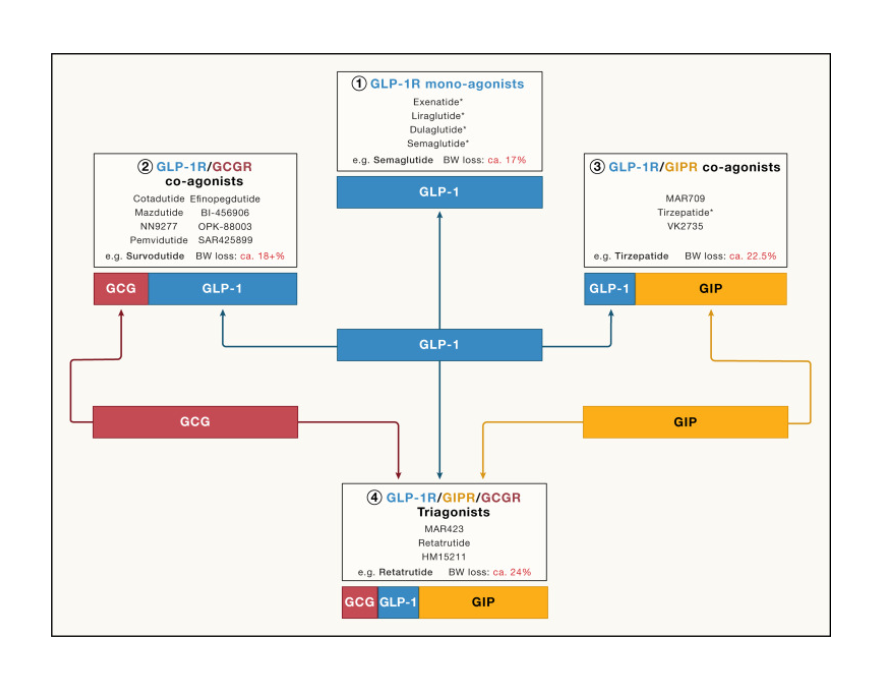
Figura 3. Agonistas multirreceptores mono y unimoleculares que han completado o se encuentran actualmente en ensayos clínicos en cursoOcultar títuloTerapéuticas basadas en péptidos basadas en (1) agonistas únicos de GLP-1R en comparación con (2) coagonistas de GLP-1R/GCGR unimoleculares, (3) coagonistas de GLP-1R/GIPR y (4) triagonistas de GLP-1R/GIPR/GCGR que han completado estudios clínicos o se encuentran actualmente en evaluación en ensayos clínicos. Los cuadros y flechas azules resaltan GLP-1, los cuadros y flechas naranjas representan GIP y los cuadros y flechas rojas muestran glucagón (GCG) y la proporción correspondiente de cada receptor hormonal en cada péptido agonista multirreceptor. Todos los coagonistas y triagonistas han mostrado efectos metabólicos beneficiosos en la reducción de los niveles de HbA1c y la reducción del peso corporal (PC) para el tratamiento de la diabetes tipo 2 y la obesidad. Se muestran ejemplos de cada categoría y sus contribuciones relativas de los respectivos ligandos, junto al porcentaje de pérdida de peso corporal (texto rojo) informado en sus ensayos clínicos correspondientes. ∗Péptidos aprobados por la FDA