Ronald Palacios Castrillo
Resumen
La interfaz cerebro-vascular-inmune ha surgido como un actor dinámico en la fisiología y la enfermedad cerebral. Akassoglou,et.al., [Cell, volumen 187, número 21,2024] proponen integrar los factores de riesgo vascular con la susceptibilidad genética como nexo para el descubrimiento de mecanismos y terapias para la neuroinflamación, la neurodegeneración y la neuroreparación en enfermedades neurológicas poligénicas.
En Detalle
La comunicación cerebro-cuerpo regida por los sistemas inmunológico y vascular ha revolucionado nuestra comprensión de la fisiología cerebral, el envejecimiento y las enfermedades neurológicas. Se han identificado mecanismos celulares y moleculares imprevistos y sitios anatómicos recientemente apreciados en los límites cerebrales. La integración de señales inmunológicas y vasculares durante la homeostasis cerebral y la enfermedad sigue siendo un área de investigación activa. Las interacciones recíprocas entre los sistemas vascular e inmunológico ocurren ya que los mecanismos inmunológicos pueden alterar la homeostasis vascular, mientras que las señales sanguíneas y vasculares también pueden instigar programas inmunológicos patogénicos robustos en el cerebro. A pesar del descubrimiento de nuevas asociaciones de genes inmunológicos y vasculares con el riesgo de enfermedad, estas variaciones genéticas son raras y no pueden predecir por sí solas la aparición y progresión de enfermedades neurológicas, que son típicamente poligénicas y esporádicas. En este comentario, Akassoglou,et.al., [Cell, volumen 187, número 21,2024] analizan los mecanismos emergentes que controlan la comunicación cerebro-cuerpo en la interfaz neurovascular y destacan la inflamación inducida por la sangre como un hilo conductor común que impulsa la patogénesis en enfermedades con diversas etiologías y predisposiciones genéticas, incluidas las enfermedades autoinmunes, infecciosas y neurodegenerativas, así como los trastornos del desarrollo neurológico. Se propone integrar la susceptibilidad genética con los desencadenantes inmunológicos y las comorbilidades vasculares como nexo para el descubrimiento de mecanismos subyacentes y nuevos objetivos terapéuticos. Finalmente, se concluye que la investigación interdisciplinaria que integra tecnologías innovadoras en la interfaz de los sistemas inmunológico y vascular marcará el comienzo de una nueva era para el descubrimiento y las terapias para las enfermedades neurológicas.
=> Recibir por Whatsapp las noticias destacadas
Integración de la susceptibilidad genética con los factores de riesgo vascular
Las enfermedades neurológicas poligénicas con una herencia modesta, como la esclerosis múltiple (EM) y la enfermedad de Alzheimer (EA), generalmente implican variantes genéticas asociadas con un mayor riesgo de enfermedad e interacciones multifacéticas entre genes y ambiente. Entre las asociaciones genéticas más significativas con las enfermedades neurológicas han surgido loci genéticos inmunes y vasculares de bajo riesgo y baja penetración. Estos loci genéticos suelen ser comunes entre otras enfermedades y no pueden predecir quién desarrollará la enfermedad. Por ejemplo, a pesar del descubrimiento de más de 250 genes asociados con la desregulación inmunológica en la EM, el inicio, la evolución de la enfermedad y la respuesta al tratamiento siguen siendo impredecibles. En gemelos idénticos, el riesgo de desarrollar EM aumenta solo en ∼30% a pesar del mismo riesgo genético entre el gemelo sano y el que tiene EM, lo que sugiere que estas variaciones genéticas pueden no ser causales y que se requiere un desencadenante adicional para el desarrollo de la enfermedad. En la enfermedad de Alzheimer de aparición tardía, varios de los loci de bajo riesgo identificados en la microglía son raros y se presentan en menos del 1 % de los pacientes. Los genes de mayor penetrancia, como APOE4, aumentan significativamente el riesgo, pero la enfermedad de Alzheimer también puede desarrollarse en personas sin el polimorfismo APOE4. La gran mayoría de los casos de muchas enfermedades neurodegenerativas son esporádicos, ya que se producen sin antecedentes familiares ni asociación causal con variantes genéticas específicas, como en la enfermedad de Alzheimer, la enfermedad de Parkinson y la esclerosis lateral amiotrófica. Los trastornos del desarrollo neurológico con etiologías complejas, como el autismo, están regidos por una multitud de mutaciones genéticas y desencadenantes ambientales que conducen a un amplio espectro de diferencias en la función cerebral. Estos hallazgos respaldan un modelo en el que se hereda un riesgo general, pero los determinantes epigenéticos adicionales y las exposiciones ambientales en última instancia dictan el inicio, la gravedad y la progresión de la enfermedad (ver Figura 1).
La disfunción vascular se ha identificado como un factor de riesgo clave entre las enfermedades neurológicas con diversas etiologías. La diabetes, la hipertensión y la cardiopatía isquémica aceleran la progresión y la gravedad de la discapacidad en la EM en 6 años. El informe de 2024 de la Comisión Lancet identificó el daño vascular y la hipertensión como los principales factores de riesgo junto con los accidentes cerebrovasculares como causas de demencia que, junto con la lesión cerebral traumática (LCT) y los desencadenantes inmunitarios periféricos, aumentan el riesgo de demencia en un 45%. De acuerdo con el informe de Lancet, la reducción del daño vascular mediante el control de la presión arterial puede mitigar el riesgo de demencia, lo que indica que la reducción del riesgo vascular puede prevenir o retrasar la demencia y conservar la cognición. El envejecimiento, las enfermedades cardiovasculares y metabólicas, la microbiota intestinal y sus metabolitos, la hipertensión y la inflamación sistémica causada por infecciones periféricas, cirugía, toxinas ambientales o cáncer pueden alterar directamente las funciones de la barrera hematoencefálica (BHE) a través de una amplia gama de mecanismos, que incluyen disfunción de los pericitos, daño de las células endoteliales, reducción del flujo sanguíneo cerebral, accidente cerebrovascular isquémico y microhemorragias. La infección por virus neurotrópicos, como el virus de Epstein-Barr (VEB), que está fuertemente vinculado con la EM, altera la integridad de la barrera hematoencefálica por efectos directos sobre las células endoteliales del cerebro. La COVID-19 puede acelerar la progresión de la demencia e inducir la alteración de la barrera hematoencefálica y la formación de coágulos sanguíneos inflamatorios, que están relacionados causalmente con la neuroinflamación y la pérdida neuronal. En los trastornos del desarrollo neurológico, la prematuridad y la hipoxia perinatal, que desencadenan hemorragia cerebral y alteración de la barrera hematoencefálica, son factores de riesgo de parálisis cerebral, discapacidad intelectual y autismo. En conjunto, estos factores de riesgo resaltan los desencadenantes vasculares e inmunitarios interconectados de las enfermedades neurológicas.
Como la disfunción vascular es causada por una amplia gama de factores de riesgo y comorbilidades con mecanismos que difieren entre enfermedades y entre pacientes, sigue siendo un desafío clave cómo abordar el daño vascular multifactorial para detener la aparición y la progresión de las enfermedades neurológicas. La extravasación de proteínas sanguíneas tóxicas en el cerebro es un punto de convergencia para los factores de riesgo vascular, independientemente del desencadenante inicial o el mecanismo que compromete la integridad de la barrera hematoencefálica (Figura 1). Estudios recientes han identificado la toxicidad de las proteínas sanguíneas en el cerebro como una vía compartida en enfermedades neurológicas con diversas etiologías vinculadas causalmente con la neuroinflamación, la neurodegeneración y la inhibición de la neuro-reparación. Se Postula que, como primer paso, la neutralización de la toxicidad sanguínea se puede aprovechar para reducir la carga del riesgo vascular en enfermedades neurológicas poligénicas con eficacia en todas las variaciones genéticas y los desencadenantes vasculares. En última instancia, el aprovechamiento de nuevos modelos computacionales y tecnologías de ácidos nucleicos para evaluar formalmente las interacciones aditivas entre los genes y los factores de riesgo vascular allanará el camino hacia terapias innovadoras y ensayos clínicos acelerados en el nexo de las vías inflamatorias y vasculares desreguladas.
La disfunción neurovascular como factor desencadenante de la activación inmunitaria
La patología neurovascular, que incluye el daño vascular o la alteración de la barrera hematoencefálica que provoca la extravasación de proteínas sanguíneas al cerebro, es un fenómeno común en las enfermedades neurológicas con diversas etiologías que se correlacionan con la aparición y progresión tempranas de la enfermedad. Las proteínas sanguíneas se estudiaron originalmente como marcadores de la alteración de la barrera hematoencefálica, y su fuga al cerebro se consideró una consecuencia de la inflamación. Los avances en imágenes, proteómica sanguínea a gran escala, perfiles multiómicos de la unidad neurovascular y estudios genéticos en modelos animales de enfermedad identificaron la interrupción de la BHE y la fuga de proteínas sanguíneas al cerebro como impulsores apicales que transforman el entorno neurovascular en un nicho proinflamatorio que promueve la neurodegeneración. Estudios transcriptómicos imparciales revelaron que las proteínas sanguíneas inducen distintos programas genéticos mediados por receptores en la microglía, destacando la influencia selectiva de los desencadenantes vasculares en las respuestas neurotóxicas de la microglía. Al unirse al receptor CD11b/CD18, el factor de coagulación fibrina induce una señalización de mecanotransducción mediada por receptores, lo que desencadena la activación inflamatoria y prooxidante de la microglía y las células inmunes periféricas (Figura 2A). Estudios farmacológicos y de pérdida de función genética identificaron que, entre las proteínas sanguíneas, la fibrina es necesaria y suficiente para la polarización neurotóxica de la microglía, mientras que su bloqueo preserva la microglía homeostática en varios modelos animales de enfermedades neurológicas, como la esclerosis múltiple, la enfermedad de Alzheimer, el traumatismo craneoencefálico y la neuropatología de la COVID-19 (Figura 2B). Esto es coherente con la reducción de la neuroinflamación mediante la inhibición de factores de la cascada de la coagulación, como el factor XII y la trombina, que son necesarios para la formación de fibrina, el producto final de la cascada de la coagulación. La neuroinflamación y la coagulación están acopladas mecanísticamente, ya que la microglía prooxidante y los macrófagos expresan factores de coagulación, que mejoran la formación de fibrina e impulsan las respuestas de las células inmunitarias innatas patógenas en tumores y enfermedades cardíacas. En consecuencia, los biomarcadores inflamatorios y de coagulación están corregulados en el envejecimiento, la esclerosis múltiple, la enfermedad de Alzheimer, la COVID prolongada y los cánceres cerebrales. Este ciclo de retroalimentación positiva crea un círculo vicioso de neuroinflamación, alteración de la BHE y un estado procoagulante que impulsa la reactividad de la microglía neurotóxica y el daño neurológico en muchas enfermedades del sistema nervioso central (SNC) con diversas etiologías.
Los desafíos que se presentan al atacar a la microglia y los macrófagos surgen al inhibir selectivamente la activación neurotóxica, preservando al mismo tiempo las funciones homeostáticas y manteniendo la vigilancia cerebral de la microglia. Las células inmunitarias innatas neurotóxicas se definen por su transición fenotípica a un estado celular reactivo que implica la liberación de moléculas tóxicas, como especies reactivas de oxígeno, que dañan las neuronas y otras células cerebrales. Estudios multiómicos y de imágenes en cerebros humanos y modelos experimentales han profundizado nuestra comprensión de la microglía, revelando nuevos conocimientos sobre su expresión genética, cambios proteómicos e interacciones complejas célula-célula con neuronas y otras células inmunes y glía durante el desarrollo, la fisiología, el envejecimiento y la enfermedad. Aunque inicialmente se caracterizaron como los fagocitos del cerebro, la microglía también juega un papel homeostático clave en la regulación de la sincronización de la red neuronal y la prevención de la hiperexcitabilidad en el cerebro sano. La microglía se polariza en células con diversas funciones efectoras, incluido el estrés oxidativo y la fagocitosis, al integrar señales externas reconocidas por una variedad de receptores celulares y proteínas. Como la fagocitosis tiene funciones protectoras, como limpiar desechos y promover la recuperación en accidentes cerebrovasculares y otras enfermedades, apuntar a poblaciones específicas de microglía y macrófagos neurotóxicos se vuelve fundamental para la . Se postula que la especificidad de los programas transcriptómicos de genes inflamatorios y prooxidantes y las vías de transducción de señales inducidas por proteínas sanguíneas se pueden aprovechar para la inactivación selectiva de respuestas inmunitarias innatas neurotóxicas. Dado que la fibrina es un desencadenante derivado de la sangre de la neuroinflamación en muchas enfermedades, puede ser un objetivo terapéutico potencial ubicuo para la inhibición selectiva de la inflamación neurotóxica (Figura 2).
Como la microglia y los macrófagos perivasculares integran continuamente señales extracelulares a través de los límites del SNC y contribuyen a la disfunción neurovascular y cognitiva, queda mucho por descubrir sobre la regulación temporal y espacial de la polarización de las células inmunitarias innatas. Los sitios de fibrillas de tau, amiloide β (Aβ), restos de mielina y depósitos de fibrina están vinculados a fenotipos moleculares y transcripcionales de la microglia asociados con la progresión de la enfermedad. De manera similar a la identificación de genes inducidos por placa alrededor de Aβ en la EA, la unidad neurovascular presenta una gran cantidad de expresión génica espacial inexplorada y cambios proteómicos a lo largo de la vida y en la enfermedad. La transcriptómica espacial con resolución de células individuales, combinada con análisis computacionales de interacciones ligando-receptor y detección in vivo con CRISPR, se puede utilizar para identificar relaciones causales entre genes, proteínas y estados celulares patológicos. Además, se pueden utilizar herramientas escalables para estudios genéticos como Perturb-seq in vivo para revelar funciones autónomas de las células e interrogar las interacciones entre células inmunitarias y vasculares en enfermedades. Como las microglias son células altamente dinámicas, la combinación de la multiómica con imágenes multimodales in vivo de dos fotones y de gran volumen corregistradas con microscopía electrónica puede proporcionar una caracterización integral de las funciones de las microglias en los límites del cerebro. Delinear el continuo de cambios transcriptómicos y estados celulares dinámicos en toda la unidad neurovascular puede revelar reguladores intrínsecos y extrínsecos imprevistos de la activación celular para la supresión selectiva de respuestas inmunitarias neurotóxicas.
Inhibición de la neuro-reparación en la interfase hematoencefálica-inmune
La inflamación y la regeneración son altamente interdependientes, y determinan el grado de daño tisular mediado por el sistema inmunitario y la posterior neuro-reparación. Estudios realizados a mediados de los años 90 mostraron que el factor de necrosis tumoral expresado en los astrocitos induce la alteración de la barrera hematoencefálica, la activación de la microglia, la infiltración de macrófagos periféricos y la muerte de oligodendrocitos, lo que induce una inflamación crónica sin posibilidad de reparación. Además de la comunicación cruzada nociva entre astrocitos y microglia, el interferón-γ (IFN-γ) , los proteoglicanos de sulfato de condroitina (CSPG) y el fibrinógeno son inhibidores extrínsecos que remodelan la matriz extracelular (ECM) e inhiben la diferenciación y la remielinización de las células progenitoras de oligodendrocitos (OPC). En conjunto, estos hallazgos resaltan los efectos directos y mediados por el sistema inmunitario de los inhibidores extrínsecos sobre la regeneración. Las propiedades estructurales y funcionales únicas de los inhibidores de la ECM de la neuro-reparación están determinadas por sus estructuras proteínicas únicas, que permiten la señalización dependiente de ligando en células inmunes, glía y células progenitoras (Figura 2A). El fibrinógeno y los CSPG se detectan en lesiones desmielinizadas en pacientes con EM y en el nicho de células madre de la zona subventricular humana después de un accidente cerebrovascular, lo que sugiere que la alteración de la BHE y la extravasación de proteínas sanguíneas, junto con factores inflamatorios, alteran el entorno extracelular para inhibir la neuro-reparación. En consecuencia, el agotamiento genético del fibrinógeno mejora la remielinización y la neurogénesis en modelos de ratón de EM, accidente cerebrovascular, trauma y lesión cerebral neonatal. Como la disfunción de la mielina es un hilo conductor común en las enfermedades autoinmunes y neurodegenerativas, las interacciones inmunitarias hematoencefálicas pueden desempeñar un papel fundamental en la neuro-reparación en todas las enfermedades. Dado que los fármacos promielinizantes actuales son insuficientes para superar la inhibición de la diferenciación de las células progenitoras neuronales por parte de los CSPG, el fibrinógeno o el IFN-γ, el descubrimiento de nuevos fármacos podría mejorar significativamente la eficacia de las terapias regenerativas en la clínica. La adición de inhibidores extrínsecos en los análisis funcionales químicos o de todo el genoma de células progenitoras neuronales, células progenitoras neuronales o células madre pluripotentes inducidas (iPSC) puede mejorar el descubrimiento de dianas y fármacos relevantes para la enfermedad con mayor potencia en las lesiones del SNC. Todavía se desconoce en gran medida si los inhibidores extrínsecos inmunitarios, sanguíneos y de la matriz extracelular utilizan programas distintos o convergen en firmas transcriptómicas compartidas para inhibir la reparación. Comprender la regulación epigenética de las células progenitoras neuronales y de oligodendrocitos expuestas a inhibidores extrínsecos podría conducir al descubrimiento de nuevos mecanismos y dianas para la neuroreparación.
Rompiendo barreras en el descubrimiento de fármacos
Las enfermedades neurológicas se han clasificado tradicionalmente en categorías distintas basadas en los mecanismos subyacentes percibidos, como neurodegenerativas, inflamatorias y vasculares. Sin embargo, el éxito limitado de los ensayos clínicos basados en estas clasificaciones tradicionales pone de relieve la necesidad urgente de reevaluar la compleja relación entre las enfermedades y sus mecanismos. La neuroinmunología está a la vanguardia de este cambio transformador en el descubrimiento de fármacos. Por ejemplo, la investigación de la inmunidad adaptativa en la EM ha dado lugar a más de 20 terapias modificadoras de la enfermedad aprobadas por la FDA para las formas recurrentes y remitentes de la enfermedad. Cabe destacar que las similitudes en los mapas de susceptibilidad de los genes inmunitarios entre la EM y la enfermedad de Parkinson indican el impacto más amplio de la inmunidad adaptativa en las enfermedades neurodegenerativas. Sin embargo, la ineficacia de los tratamientos actuales para la EM progresiva y otras enfermedades neurodegenerativas con disfunción inmunitaria innata y patología vascular prominentes subraya la necesidad de nuevas terapias para combatir la neurodegeneración.
La investigación del paisaje vascular del SNC ya muestra resultados prometedores para nuevas estrategias terapéuticas. Neutralizar la toxicidad sanguínea al atacar la fibrina con un anticuerpo monoclonal inhibe selectivamente las respuestas inmunitarias neurotóxicas y actualmente se encuentra en desarrollo clínico (Figura 2B). La selección de la fibrina como objetivo terapéutico se basó en estudios de investigación clínica y básica que determinaron la causalidad, la selectividad y la relevancia para la enfermedad humana por parte de múltiples laboratorios en enfermedades neurológicas y periféricas durante los últimos 30 años. Se desarrolló una inmunoterapia de primera clase dirigida a un epítopo críptico de fibrina para inhibir selectivamente la inflamación sin efectos anticoagulantes adversos ni inmunosupresión general. El anticuerpo de fibrina inhibe selectivamente las vías inmunitarias innatas neurotóxicas con eficacia en modelos de ratones con EM, EA y COVID-19 y actualmente se está probando en ensayos de fase 1. Dirigirse a la fibrina puede ser prometedor para neutralizar la patología inmunitaria inducida por vasos sanguíneos compartida en enfermedades autoinmunes y neurodegenerativas y traumatismos, también en combinación con otros tratamientos que eliminen Aβ o que se dirijan a la inmunidad adaptativa. Las vías compartidas que controlan la integridad de la BHE, como la vía Wnt/β-catenina que mitiga la disfunción endotelial, también se investigan por su potencial terapéutico para restablecer las funciones de la BHE. La plataforma experimental de fibrina (Figura 2) puede servir como guía para que otras proteínas sanguíneas y dianas vasculares descubran sus mecanismos celulares y moleculares en el SNC, prueben la causalidad con estudios genéticos y desarrollen terapias para ensayos clínicos.
Un desafío clave sigue siendo cómo se seleccionarán, validarán, probarán y reducirán los riesgos de manera acelerada nuevos objetivos neuroinmunes y vasculares. Varias vías inmunitarias son prescindibles o tienen funciones pleiotrópicas y protectoras, lo que limita su utilidad como dianas terapéuticas. Los ensayos clínicos en curso que se dirigen a loci genéticos de bajo riesgo y baja penetrancia revelarán si pueden ser eficaces como monoterapias en enfermedades neurológicas poligénicas o terapias combinadas para proteger de cambios epigenéticos y comorbilidades. Además, considerar los efectos potenciales del riesgo genético en el desarrollo de comorbilidades vasculares sistémicas puede proporcionar una nueva comprensión de la comunicación cerebro-cuerpo y mejorar la estratificación de los pacientes para ensayos clínicos. El diseño de nuevos modelos animales de enfermedad se beneficiaría de la integración de variaciones genéticas con desencadenantes inmunitarios sistémicos y factores de riesgo vascular. Dada la heterogeneidad de las enfermedades neurológicas, un enfoque integrado que consista en conjuntos de datos epidemiológicos a gran escala, biomarcadores, neuropatología, multiómica y respuesta a los tratamientos puede proporcionar un modelo unificador que reconozca la complejidad de las interacciones no lineales entre las vías biológicas. El aprendizaje automático se puede aprovechar para predecir la aparición de la enfermedad, la progresión y la respuesta a los tratamientos. Dentro de este contexto integrado, surgirán nuevos objetivos en la intersección de las vías entre la susceptibilidad genética, los determinantes epigenéticos y los desencadenantes vasculares. En conjunto, estos enfoques crearán una innovadora línea de productos biológicos, moléculas pequeñas y terapias celulares que se están probando en ensayos clínicos para enfermedades neurológicas que actualmente no reciben suficiente atención o que no tienen tratamiento.
Conclusiones
La inmunología neurovascular del cerebro ha anunciado el advenimiento de una investigación multidisciplinaria que integra la inmunología y la neurociencia con la biología vascular y la hematología para estudiar la interfaz sangre-cerebro-inmunidad. Aprovechando los avances en genómica, imágenes y aprendizaje automático, este campo emergente está preparado para revolucionar nuestra comprensión de la fisiología y la enfermedad cerebral con descubrimientos que cambiarán paradigmas a un ritmo sin precedentes. Los estudios preclínicos rigurosos y el desarrollo de modelos experimentales de vías relevantes para la enfermedad y exposiciones ambientales podrían ser fundamentales para diseccionar los mecanismos neurovasculares en la enfermedad. Desentrañar la causalidad de los genes inmunes y vasculares, los cambios epigenéticos y los desencadenantes ambientales asociados con la aparición y la progresión de la enfermedad es muy prometedor para el desarrollo de nuevas terapias. En última instancia, la integración de las puntuaciones de riesgo poligénico con los aceleradores de enfermedades ambientales y las variables clínicas será fundamental para marcar el comienzo de una nueva era de terapias inmunitarias y vasculares de precisión para las enfermedades del sistema nervioso central. En la próxima década, los avances científicos en la interfaz sangre-cerebro-sistema inmunitario surgirán de redes colaborativas interdisciplinarias de inmunólogos, neurocientíficos, hematólogos, genetistas, informáticos, físicos, bioingenieros, desarrolladores de fármacos e investigadores clínicos. Estas asociaciones entre el mundo académico, la industria, las fundaciones y las empresas catalizarán la innovación en el descubrimiento de fármacos y transformarán la práctica médica para las enfermedades neurológicas.
Una amplia gama de factores de riesgo vascular y comorbilidades inducen la extravasación de proteínas sanguíneas hacia el SNC, lo que es un hilo conductor común en enfermedades neurológicas con diversas etiologías que impulsan respuestas inmunitarias en el SNC. La patología vascular, la alteración de la barrera hematoencefálica, los factores sanguíneos tóxicos y la coagulación están interconectados con la neuroinflamación y son mecanismos compartidos entre enfermedades con diversas etiologías, como la esclerosis múltiple, la enfermedad de Alzheimer, la COVID-19, los accidentes cerebrovasculares y los traumatismos craneoencefálicos. La susceptibilidad genética y las comorbilidades apuntan a orígenes inmunitarios y vasculares de enfermedades neurológicas poligénicas e interacciones aditivas entre genes y medio ambiente. Los enfoques experimentales integrados para estudiar la interfaz neurovascular pueden proporcionar un modelo unificador de los factores de riesgo ambientales y las predisposiciones genéticas y revelar nuevos objetivos terapéuticos.
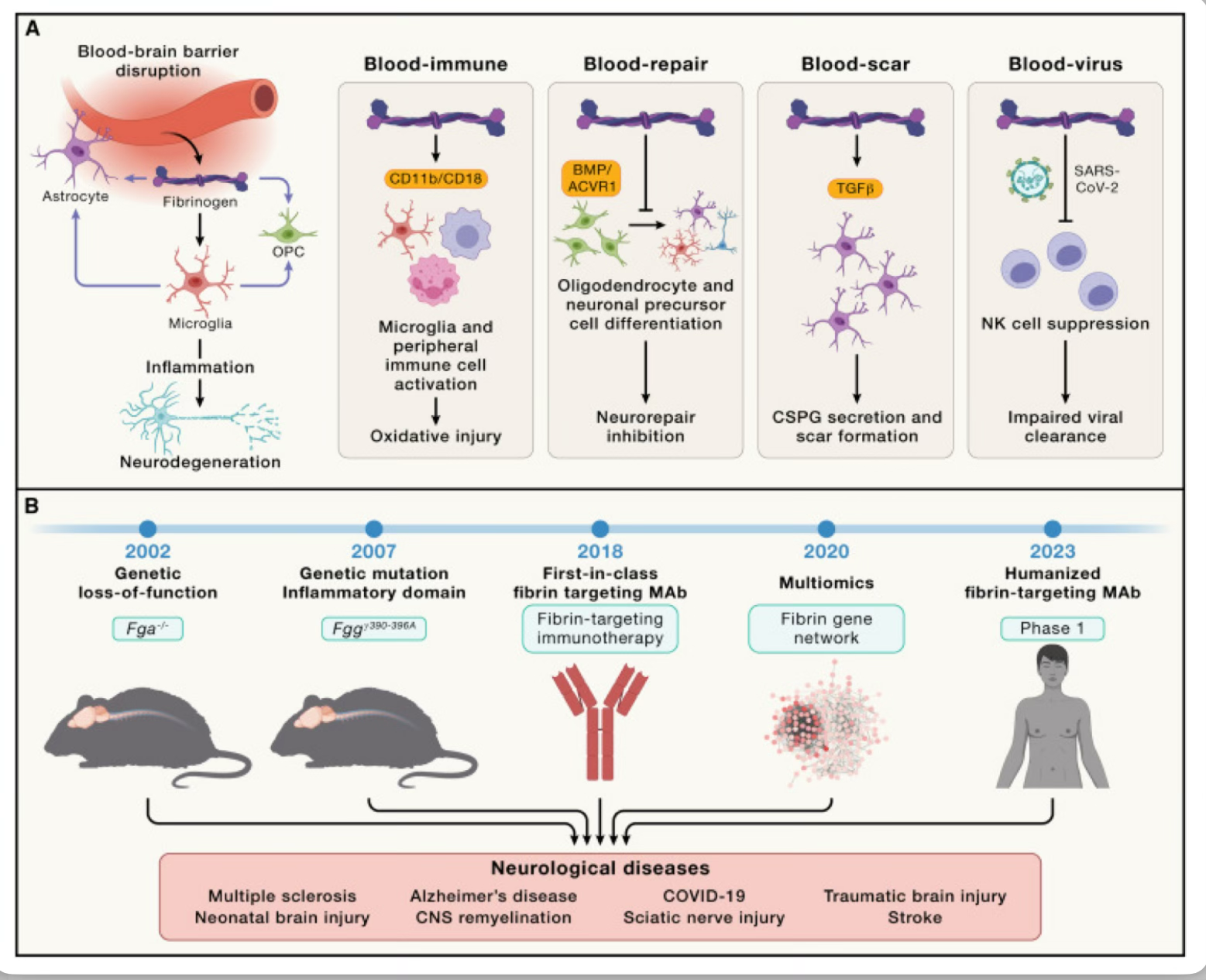
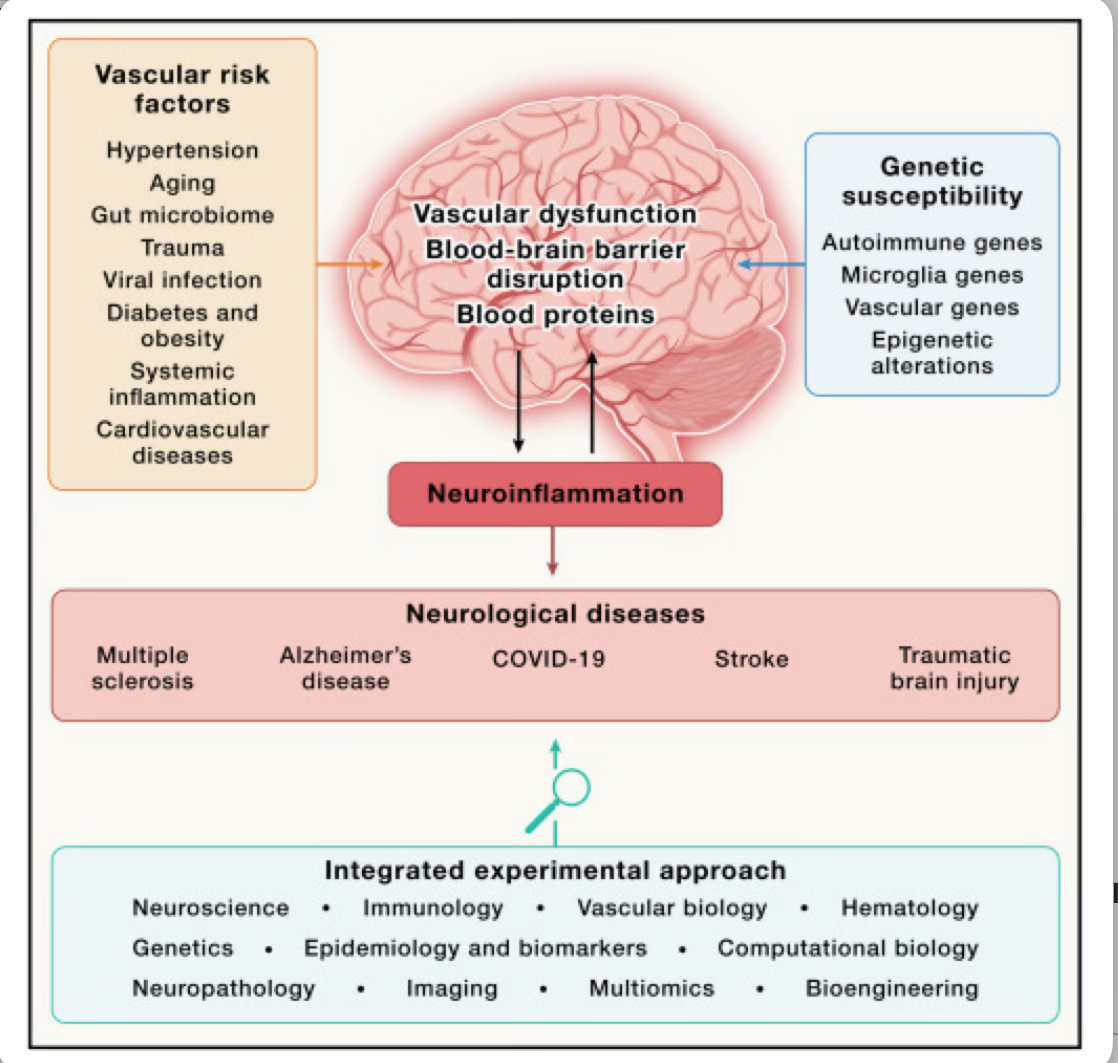
Figura 2.
- Mecanismos y funciones de la fibrina en la neuroinflamación y la neuroreparación. El fibrinógeno se extravasa en el SNC tras la interrupción de la barrera hematoencefálica, donde se convierte en fibrina tras la activación de la coagulación La fibrina, a través de las integrinas CD11b/CD18, activa la microglia y las células inmunitarias innatas para inducir la inflamación patógena y la lesión oxidativa, lo que conduce a la neurodegeneración. Mediante la activación de la señalización del receptor de la proteína morfogenética ósea (BMP)/receptor de activina A tipo 1 (ACVR1), el fibrinógeno bloquea la diferenciación de oligodendrocitos y células precursoras neuronales, inhibiendo la neuroreparación. Como portador del factor de crecimiento transformante β (TGFβ) latente, el fibrinógeno induce la secreción de CSPG y la formación de cicatrices en los astrocitos. En la COVID-19, la fibrina interactúa con la proteína Spike del SARS-CoV-2, lo que induce coágulos anormales con una actividad proinflamatoria aumentada y suprime las células NK y la eliminación viral. (B) Estudios de pérdida de función genética y desarrollo de inmunoterapia con fibrina. Ratones knock-out de fibrinógeno (Fga−/−) y Fggγ390-396A, que expresan fibrinógeno mutante que conserva la función de coagulación normal pero carece del motivo de unión del fibrinógeno γ390-396 a CD11b/CD18, mostraron protección en modelos de EM, EA, LCT, lesión cerebral neonatal y COVID-19. Una inmunoterapia con fibrina de primera clase dirigida al epítopo inflamatorio de fibrina γ377-395 bloquea selectivamente la inflamación sin efectos adversos en la coagulación. La transcriptómica y la fosfoproteómica identificaron la firma única de fibrina en las células inmunes innatas y la selectividad que apunta a la fibrina para inhibir la inflamación neurotóxica. La inmunoterapia con fibrina protege contra la desmielinización inflamatoria y el daño axonal en modelos animales de EM, reduce la neurodegeneración y las redes de genes inflamatorios neurotóxicos en la corteza de ratones con enfermedad de Alzheimer, y protege contra la neuroinflamación y la pérdida neuronal en modelos de infección de COVID-19. Actualmente, se está probando una versión humanizada de la inmunoterapia con fibrina en ensayos de seguridad de fase 1 en voluntarios sanos. Esta línea experimental que combina la biología celular y molecular, la pérdida de función genética y los estudios multiómicos en modelos animales de enfermedades neurológicas, el desarrollo de terapias de primera clase y las pruebas clínicas se puede utilizar para descubrir mecanismos de acción, probar la causalidad y la selectividad, y desarrollar terapias para otras proteínas sanguíneas y objetivos vasculares en la interfaz neurovascular.