Introducción
Las enfermedades neurodegenerativas (NDD) son un grupo heterogéneo de trastornos neurológicos que afectan negativamente la vida de millones de personas en todo el mundo y conllevan la pérdida progresiva de neuronas en el sistema nervioso central (SNC) o el sistema nervioso periférico (SNP). El colapso de la estructura y función de las redes neuronales y la pérdida de neuronas, que no pueden renovarse eficientemente debido a su naturaleza terminalmente diferenciada, dan como resultado la ruptura del circuito comunicativo central, que culmina en deterioro de la memoria, la cognición, el comportamiento, sensorial, y/o función motora.
En esta revisión, se argumenta que un conjunto de características definen los NDD, a saber: agregación patológica de proteínas, disfunción de la red neuronal y sináptica, proteostasis aberrante, anomalías del citoesqueleto, metabolismo energético alterado, defectos de ADN y ARN, inflamación y muerte de células neuronales (Figura 1). ).
Se describen estas características y su evidencia en el contexto de los NDD prevalentes, incluida la enfermedad de Alzheimer (AE), la enfermedad de Parkinson (PD), tauopatías primarias , demencia frontotemporal (FTD), esclerosis lateral amiotrófica (ELA), sinucleinopatías (es decir, demencia con cuerpos de Lewy [LBD] y atrofia multisistémica [MSA]) , enfermedad de Huntington (HD) y enfermedades relacionadas con la poliglutamina (polyQ) (incluidas las ataxias espinocerebelosas [SCA]), enfermedad priónica (PrD), lesión cerebral traumática (TBI), encefalopatía traumática crónica (CTE), accidente cerebrovascular, lesión de la médula espinal (SML) y esclerosis múltiple (MS).
=> Recibir por Whatsapp las noticias destacadas
La epidemiología, los síntomas, la genética y las firmas patológicas de estos NDD específicos se han y ampliamente revisado previamente , (Figura 2). En este trabajo, se definen un sello distintivo de NDD como un proceso celular o molecular que cumple con los siguientes criterios: (1) está vinculado a formas genéticas (raras) de NDD, (2) contribuye a formas esporádicas de NDD, (3) contribuye a neurodegeneración y pérdida neuronal en modelos preclínicos y pacientes con NDD, y (4) se alteran los marcadores moleculares que reflejan el sello distintivo.
Además de describir las características y la evidencia genética y biológica de estas características, se destaca las interrelaciones de los procesos celulares y moleculares subyacentes y cómo las características pueden detectarse y monitorearse usando biomarcadores in vivo. Se propone que el proceso neurodegenerativo en NDD está impulsado por defectos combinados en múltiples características de NDD, lo que apunta a la necesidad de terapias con objetivos múltiples. Las características primarias o principales del NDD que impulsan un NDD específico en un individuo dependerán del insulto del NDD y de la vulnerabilidad y resiliencia neuronal, es decir, la capacidad de manejar y protegerse contra los insultos, del individuo y la región del cerebro afectada.
Aquí se presenta un marco para estudiar los NDD utilizando un enfoque holístico que involucra la interconexión y la participación combinada de múltiples características en el proceso neurodegenerativo. Este marco general se puede utilizar para comprender los mecanismos moleculares de la neurodegeneración, para categorizar diferentes NDD en función de las características principales, para estratificar subtipos y pacientes dentro de NDD específicos en función de las características principales y para diseñar estrategias terapéuticas combinatorias o personalizadas para detener de manera efectiva los NDD. .
Agregación patológica de proteínas
Los agregados patológicos de proteínas son características de los NDD.
La agregación característica de proteínas es un sello patológico clave de una variedad de NDD y, a menudo, sirve para el diagnóstico y la clasificación de enfermedades (Figuras 1 y 3). Estos NDD se clasifican como proteinopatías e incluyen: AD, PD, primaria tauopatías (incluyendo parálisis supranuclear progresiva [PSP], degeneración corticobasal [CBD], demencia frontotemporal ligada a tau [FTD-tau]), FTD, ALS, sinucleinopatías (incluyendo LBD y MSA), HD y enfermedades poliQ relacionadas (incluyendo SCA), y PrD. La proteína de agregación característica, los genes vinculados, los síntomas y las regiones cerebrales afectadas de estos diferentes NDD se presentan en la Figura 2 y 3 . En particular, para muchos NDD, los agregados de proteínas se encuentran en regiones del cerebro que se correlacionan con los síntomas clínicos, lo que respalda su papel patogénico en los NDD, esto apoya que la vulnerabilidad neuronal selectiva debe considerarse (Figura 2).
Conocimientos mecanicistas de la genética: ganancia de función tóxica versus pérdida de función
La identificación de mutaciones causales para formas hereditarias (raras) de NDD ha ayudado a la búsqueda de mecanismos patogénicos de NDD, es una característica común entre diferentes NDD, lo que respalda un mecanismo tóxico de ganancia de función. (AD y tauopatías), α-sinucleína (PD y sinucleinopatías), PrPC (PrD), SOD1 (ALS/FTD), TAR DNA-binding protein 43 (TDP-43) (ALS/FTD), FUS (ALS/FTD) , y huntingtina (Htt) (HD). El hecho de que las mutaciones en diferentes NDD mejoren la agregación de la proteína NDD característica apunta a un papel patógeno central para agregación de proteínas en los NDD ,como Presenilin 1 y 2 (PSEN1, PSEN2) que no conducen a la agregación de la proteína codificada, sino que aumentan la agregación de proteínas NDD clave. Otro mecanismo de ganancia de función es la generación de repeticiones de dipéptidos agregantes ( DPR) de secuencias repetidas de hexanucleótidos, generadas por traducción no ATG (RAN) asociada a repetición. La estrecha correlación entre el proceso de agregación y la progresión de los síntomas en la mayoría de los NDD y el hecho de que las proteínas afectan las funciones neuronales cruciales y las características de NDD (descritas a continuación) respaldan aún más una ganancia tóxica de función.
La función normal de los genes vinculados a los NDD también está relacionada con diferentes características de los NDD (por ejemplo, para tau, APP, α-sinucleína, SOD1, FUS,y TDP-43 [descrito a continuación]). Además, la agregación y el secuestro de proteínas en una parte de la célula pueden provocar la pérdida de su presencia en otra y, por tanto, la pérdida de su función fisiológica. Los efectos combinados de la ganancia tóxica y la pérdida de función podrían conducir a defectos concomitantes en las características del NDD o a un proceso de múltiples impactos que impulsa la neurodegeneración (Figuras 1 y 3).
Propagación similar a priones
En la PrD, el plegamiento incorrecto de la proteína priónica y la rápida propagación de su agregación se ha identificado como el principal mecanismo responsable de propagar la neurodegeneración entre las células y las regiones del cerebro. La pérdida rápida y dramática de las neuronas y la función de la red en la PrD proporciona un argumento convincente para la agregación de proteínas y su propagación como un impulsor importante del proceso neurodegenerativo y, por lo tanto, como un sello distintivo de NDD (Figuras 1 y 3). Este mecanismo proporcionó un concepto novedoso tanto para los PrD como para otros NDD por priones verdaderos ya que no hay evidencia de transferencia entre individuos. El concepto similar al prión también condujo a la identificación de diferentes cepas de elementos similares al prión en los NDD, que se correlacionan con la toxicidad y la propensión a la agregación y la propagación. En consecuencia, los procesos similares a los priones se consideran contribuyentes a la neurodegeneración y la progresión espacio-temporal característica de la patología en los NDD proteinopáticos, aunque queda por considerar la vulnerabilidad neuronal selectiva (Figuras 1 y 3).
Agregación de proteínas y toxicidad.
Aunque la agregación de proteínas se correlaciona en gran medida con la progresión de los síntomas en los NDD proteinopáticos, esto no implica directamente que los agregados maduros sean los principales culpables tóxicos. En particular, se han propuesto ensamblajes oligoméricos intermedios como candidatos neurotóxicos. Tanto los agregados fibrilares maduros como los ensamblajes oligoméricos están presentes extra o intracelularmente, en diferentes tipos de células y en diferentes ubicaciones subcelulares según la proteína involucrada (es decir, nuclear, citoplasmática, y presináptica o postsináptica), lo que permite su interferencia con diferentes características de NDD (descritas a continuación), (Figuras 1 y 3), aunque el mecanismo tóxico exacto y las formas tóxicas de estos agregados aún no han sido definidos unívocamente. Es de destacar que los agregados de proteínas no siempre se correlacionan perfectamente con el proceso de la enfermedad. Por ejemplo, para algunos DPR, la correlación con la progresión de la enfermedad es bastante débil, y en algunos casos genéticos (p. ej., ligado al gene de la quinasa 2 repetida rica en leucina [LRRK2] o al gene Parkin [PRKN]) no se observan agregados de proteínas fibrilares. Estos hallazgos sugieren que en estos casos se deben considerar mecanismos alternativos para la neurotoxicidad y que los eventos moleculares distintos de las proteínas agregadas contribuyen a la enfermedad.
Biomarcadores para la agregación de proteínas NDD
Se han desarrollado varios ensayos de biomarcadores bien caracterizados para la agregación de proteínas con valor diagnóstico para NDD. Por ejemplo, la tomografía por emisión de positrones (PET) de amiloide y la proporción de péptidos Aβ (Aβ42/Aβ40) de 42 a 40 aminoácidos de longitud en el líquido cefalorraquídeo (LCR) se pueden utilizar para monitorear el depósito de amiloide en el contexto de la AD. De hecho, ambos ensayos detectan el inicio de la agregación de Aβ décadas antes del inicio de la enfermedad clínica. El plasma Aβ42/Aβ40 también se puede usar para detectar la patología temprana de Aβ, aunque muestra diferencias absolutas más bajas que el LCR.
Marcadores adicionales que reflejan la acumulación de amiloide
En el cerebro se encuentran formas de tau fosforiladas en LCR y plasma, lo que refleja un vínculo entre ambos procesos patológicos en la AD. El biomarcador mejor establecido para la patología de tau en la AD es tau-PET. Es importante destacar que tau-PET es menos adecuado para las taupatías primarias y las taupatías secundarias distintas de la AD (p. ej., CTE) que para la tauopatía de la AD, debido a las diferencias conformacionales. Para TDP-43, α-sinucleína o agregados de priones, ha sido más difícil desarrollar biomarcadores de fluidos clásicos que sean específicos de la patología. Sin embargo, el hecho de que estas proteínas puedan propagarse de manera similar a un prión provocó la idea de que se podrían usar ensayos de agregación de siembra, como la conversión inducida por temblores en tiempo real (RT-QuIC) o la amplificación cíclica de plegamiento incorrecto de proteínas (PMCA) para detectar cualitativamente formas patológicas de las proteínas en LCR. Los estudios que analizan el LCR lumbar con RT-QuIC o PMCA de proteína priónica y α-sinucleína se han convertido en pruebas clínicas con excelente precisión diagnóstica y confirmación neuropatológica.
Trastornos neurodegenerativos no proteinopáticos
Con base en la evidencia anterior, la agregación de proteínas se considera un contribuyente importante al proceso neurodegenerativo en los NDD proteinopáticos. Sin embargo, el papel de la agregación de proteínas en los NDD con un componente traumático, isquémico o inflamatorio primario puede ser menos claro. Dichas enfermedades no proteinopáticas incluyen TBI, CTE, accidente cerebrovascular, SCI, y MS, donde el insulto primario no está obviamente relacionado con la agregación de proteínas. Sin embargo, varios de estos NDD muestran agregación de proteínas como un presunto efecto secundario, lo que contribuye a una fase agravante crónica (p. ej., tau y TDP-43 en TBI y CTE, tau y neurofilamento en MS, y tau en SCI). También hay NDD genéticos en los que no se observa proteinopatía, por ejemplo, atrofia muscular espinal (SMA), parkinsonismo recesivo y algunos casos de PA genéticos (por ejemplo, LRRK2). Por lo tanto, no siempre podemos asignar un papel principal a la agregación de proteínas en la patogénesis de todos los NDD, pero la agregación de proteínas puede contribuir a la progresión de la enfermedad en combinación con otras características de los NDD.
Disfunción de las redes sinápticas y neuronales
En los NDD, los síntomas típicamente reflejan la alteración de redes neuronales específicas, y la falla sináptica y la toxicidad parecen ser un evento temprano que precede a la pérdida neuronal en muchos NDD (Figuras 1 y 3). La función de la red neuronal requiere una función sináptica precisa, así como una regulación controlada de la estabilización y eliminación de sinapsis. La función sináptica, a su vez, está modulada por cambios de neurotransmisores y calcio, adaptaciones del citoesqueleto, dinámica de vesículas presinápticas y señalización postsináptica (Figura 3). La función sináptica requiere una regulación estricta de la función mitocondrial y el suministro de energía para mantener la homeostasis del calcio y el equilibrio iónico, incluso mediante bombas de membrana que restablecen los gradientes de iones durante la señalización neuronal. También se requiere energía para la eliminación y reposición de constituyentes para función sináptica adecuada, que exige transporte axonal estrechamente controlado y coordinado, dinámica del citoesqueleto, proteostasis, metabolismo de lípidos y RNA, autofagia, y homeostasis mitocondrial. Además, los astrocitos y la microglía desempeñan importantes funciones autónomas no celulares en la energía y homeostasis de neurotransmisores, eliminación y estabilización de sinapsis (Figura 3).
Varias revisiones destacan la evidencia genética, preclínica y derivada del paciente que respalda un papel clave de la falla y disfunción sináptica en los NDD. Por ejemplo, la disfunción sináptica (es decir, hiperexcitación o excitotoxicidad) combinada con dishomeostasis del calcio y agotamiento de la energía juega un papel clave en el accidente cerebrovascular por exceso de entrada de Ca2+. El aumento de Ca2+ provoca disfunción mitocondrial y agotamiento energético concomitante, así como activación de enzimas, como las calpaínas, lo que resulta en la degradación de proteínas y lípidos, desregulación de las funciones fisiológicas y, en última instancia, muerte celular. La hiperexcitabilidad neuronal y la excitotoxicidad mediada por glutamato también se han considerado mecanismos importantes en la etiología de la ELA y pueden contribuir al proceso neurodegenerativo en HD, AD, MS, SCI,y TBI también. Además, varias proteínas que agregan en los NDD ejercen un papel fisiológico en la sinapsis y/o las formas patológicas asociadas inducen una falla o disfunción sináptica, lo que subraya la interconexión de las características distintivas de los NDD (p. /Aβ y tau en AD y tauopatías). Además, se sabe que varios genes que están relacionados con la función sináptica adecuada están mutados en ciertos NDD (por ejemplo, SNCA, SYNJ, DNAJC y DNAJC en PD, y C9orf72121 en ALS/FTD). Por lo tanto, es probable que se observe una combinación de sinapsis hipo e hiperactivas en los NDD, lo que provoca un patrón complejo de neurotransmisión desregulada en el cerebro.
La falla y la disfunción sináptica en los NDD se han descrito como un evento temprano antes de la neurodegeneración en AD, PD, HD, FTD, ALS y accidente cerebrovascular.Por ejemplo, la falla sináptica y la pérdida de sinapsis en la AD ocurrieron antes de la pérdida neuronal, mientras que también se mostró hiperexcitabilidad en diferentes regiones del cerebro. De manera similar, la descomposición sináptica y axonal precede a la pérdida neuronal en la PD, y al inicio de los síntomas, la sinapsis la disfunción supera la pérdida de neuronas dopaminérgicas. También se encontró que los síntomas y los defectos sinápticos preceden a la pérdida neuronal manifiesta en la HD y la desconexión entre las neuronas motoras y el músculo se observa antes de que las neuronas motoras mueran en la ELA. Apoyando aún más el papel de los defectos sinápticos en los NDD, la expresión de mutantes patogénicos, como APP, tau, α-sinucleína, Htt, y varios genes ALS/FTD, induce una disfunción de la red neuronal y sináptica en modelos preclínicos y se asocia espacial y temporalmente con agregados proteicos tempranos , particularmente agregados oligoméricos. La agregación de varias proteínas NDD puede ocurrir dentro de las sinapsis, ya sea en la especialización presináptica o postsináptica, y está asociada con efectos adversos en la función sináptica.
La mejora sintomática de los NDD después de la administración de medicamentos basados en la modulación de la neurotransmisión sugiere un papel para la función sináptica. Un ejemplo poderoso de la modulación de la neurotransmisión es el reemplazo de la señal de dopamina perdida en PD utilizando el precursor L-DOPA, que es temporalmente efectivo para restaurar los síntomas motores en la enfermedad temprana a moderada , la mejoría sintomática leve durante varios meses después de la administración de inhibidores de la acetilcolinesterasa y antagonistas de NMDAR en la SA, riluzol, que modula la neurotransmisión glutamatérgica, en la ELA y tetrabenazina que modula la señalización dopaminérgica en la corea del HD.
Los defectos sinápticos se pueden detectar mediante una serie de modalidades de imagen en pacientes con NDD. La resonancia magnética funcional (fMRI), que evalúa la conectividad de la red neuronal, indica una disfunción temprana de la red en los NDD. La fluorodesoxiglucosa-PET (FDG-PET) revela una disminución del metabolismo de la glucosa en la AD, en consonancia con la disminución de la actividad neuronal en la enfermedad. Además, recientemente se desarrolló un método de PET que obtiene imágenes de la densidad sináptica utilizando un ligando dirigido a la proteína de la vesícula sináptica 2A (SV2A), y ahora también hay ensayos dirigidos para medir paneles de proteínas presinápticas, trans y postsinápticas en el LCR, con resultados prometedores en todos los NDD.
La disfunción de la red sináptica y neuronal interactúa estrechamente con otras características del NDD (Figura 3). Como se describió anteriormente, la función correcta de la red sináptica y neuronal requiere el funcionamiento correcto y la interacción con los procesos fisiológicos centrales relacionados con las características del NDD. Como tal, la disfunción sináptica y la excitotoxicidad están estrechamente relacionadas con el metabolismo energético defectuoso, el estrés oxidativo, la producción de proteínas (locales) por el RNA transportado por los axones, la degradación de proteínas y organelos, la dinámica del citoesqueleto y la muerte neuronal. La sinapsis puede actuar como un iniciador celular autónomo de la disfunción y muerte celular, dependiendo de la vulnerabilidad y resistencia de la neurona. Además, los procesos autónomos no celulares pueden contribuir al daño sináptico. Por ejemplo, la activación microglial puede conducir a una poda sináptica inapropiada, puede contribuir a la propagación de proteínas mal plegadas en la sinapsis y puede afectar las interacciones microgliales/astrocíticas que son clave para la función de la sinapsis.
Proteostasis aberrante
La acumulación de proteínas agregadas ubiquitinadas en muchos NDD (p. ej., tau, TDP-43, α-sinucleína, Htt,…), así como la presencia de p62 en agregados de NDD, indica proteostasis alterada en NDD; la evidencia acumulada implica aún más la proteostasis aberrante en los NDD (Figuras 1 y 3). El sistema de ubiquitina-proteasoma (UPS) y la vía de autofagia-lisosoma (ALP) constituyen dos mecanismos celulares principales para mantener la homeostasis de las proteínas por mitofagia, a través de la absorción de material celular por una estructura de doble membrana, el autofagosoma. La UPS y la ALP implican ubiquitilación para apuntar a proteínas y carga para la degradación y están unidas por p62 (SQSTM1), que se une a la ubiquitina y dirige la carga a el ALP, con la fusión de vesículas autofágicas y la subsiguiente degradación lisosomal siendo el paso final. Son inducidas por condiciones de inanición o estrés que involucran baja energía o baja disponibilidad de constituyentes (a saber, aminoácidos específicos) y luego de la eliminación de proteínas y orgánelos dañados en el citosol proporciona nuevos constituyentes para la síntesis de macromoléculas y sustratos energéticos, destacando su interconexión con otras características distintivas de NDD. La ALP también actúa en el axón y la sinapsis, donde contribuye a la homeostasis local de proteínas y la mitofagia que se requieren para una función sináptica eficaz. La autofagia y la función lisosomal desreguladas están estrechamente relacionadas con las vías de muerte celular, lo que lleva a la muerte neuronal.
El sistema ubiquitina–proteosoma
La observación de que varios genes vinculados a formas familiares de NDD mantienen funciones clave en la respuesta del UPS respaldan un papel activo del UPS en los NDD. , están asociados con ALS/FTD. Además, Parkin (un gene ligado al PD) codifica una ubiquitina-proteína ligasa, y UCHL1, vinculado a una rara forma progresiva de NDD, codifica una hidrolasa ubiquitina carboxi-terminal, lo que apunta aún más a un papel del UPS en los NDD. Además, la agregación de proteínas asociadas con NDD esporádicos, como tau, TDP-43, α-sinucleína y proteínas que contienen poliQ (polyglutaminas), altera la función del UPS, lo que sugiere una asociación más amplia con formas más comunes de NDD. Proteínas de choque térmico, incluidas HSP70 y HSP90 , que son actores clave para facilitar el plegamiento de proteínas, se ha demostrado que modulan el recambio o la estabilización de Aβ mal plegado, tau, α-sinucleína, y TDP-43. Finalmente, la agregación de proteínas se ha relacionado con niveles reducidos de ATP, reuniendo la agregación de proteínas y el metabolismo de la energía neuronal (discutido más adelante), enfatizando las posibles interacciones exacerbantes entre las diferentes características del NDD.
La vía lisosomal de la autofagia
De acuerdo con que la autofagia es crítica para la salud neuronal, la deficiencia de algunos genes atg, que se sabe que son críticos para la regulación de la autofagia, causa neurodegeneración. Como tal, la inactivación específica del cerebro de la autofagia por la desactivación de Atg7 en ratones causa neurodegeneración y muerte prematura con acumulación concomitante de proteínas agregadas. De manera similar, la deficiencia neuronal posnatal de Atg5 da como resultado neurodegeneración y acumulación de proteínas agregadas en ratones. La autofagia mediada por chaperonas también previene el colapso del proteoma metaestable neuronal que incluye proteínas relacionadas con la AD. Además, varios genes vinculados a los NDD ejercen funciones fisiológicas en la regulación de la autofagia. Mutaciones en el gene SQSTM1 que codifica la proteína p62, que une UPS y ALP al unirse a la ubiquitina y dirigir la carga a la ALP, dan lugar a casos de ALS/FTD. Los genes asociados con PD ejercen funciones cruciales en la autofagia, la función lisosomal y el tráfico endolisosomal, incluidas varias proteínas clave (es decir, LRRK2, SYNJ1, DNAJC6 y DNAJC13 [ RME-8]) importante para controlar la endocitosis y la autofagia en la sinapsis. De manera similar, Los genes asociados con los NDD codifican proteínas (es decir, PINK1, Parkin, LRRK2, α-sinucleína, Htt y ataxina 3) que están involucradas en la autofagia, el tráfico endolisosomal y la degradación de proteínas. La ALP también actúa a nivel de la sinapsis, contribuyendo a la homeostasis de proteínas sinápticas y, en consecuencia, a una función sináptica precisa. Además, se han detectado proteínas de agregación en sitios presinápticos y postsinápticos donde pueden causar desregulación de la ALP y la función sináptica.
La evidencia del papel de la disfunción lisosomal en la neurodegeneración se enfatiza además en los trastornos de almacenamiento lisosomal (LSD). Existe una gran familia de LSD donde la neurodegeneración es parte de la presentación clínica de la enfermedad debido a mutaciones recesivas de pérdida de función. Estas formas neuropáticas de inicio temprano de la enfermedad incluyen, entre otras, la enfermedad de Niemann-Pick tipo C1 (NPC) y la enfermedad de Gaucher, que están relacionadas con defectos lisosomales que dan lugar a una alteración de la homeostasis del colesterol o de los lípidos, respectivamente. Curiosamente, existe un sorprendente paralelo en algunos de los síntomas y las proteínas de agregación de los LSD con ciertos NDD (p. ej., tau en NPC). Por el contrario, algunos genes asociados con los NDD están vinculados a los LSD, como GBA (β-glucocerebrosidasa) y GRN (progranulina). Los genes asociados con los NDD ejercen funciones lisosomales clave (por ejemplo, ATP13A2 [exportador de poliamina lisosomal] en la enfermedad de Parkinson).
La agregación de proteínas afecta negativamente la función de ALP, y la ALP también parece desempeñar un papel en la transferencia de célula a célula de proteínas propensas a la agregación, similar a la propagación similar a priones discutida anteriormente. Por ejemplo, el tráfico endolisosomal que sigue a la absorción de tau y la ruptura lisosomal posterior promueve el acceso citosólico a tau agregada. Además, las fibrillas de α-sinucleína que se introducen en las células tienen efectos disruptivos sobre la función lisosomal.Se han desarrollado paneles de biomarcadores para proteínas lisosomales y se han observado concentraciones alteradas tanto en la PD como en la AD, pero se necesitan más estudios para su comprensión detallada y su uso posterior.
La disfunción lisosomal y autofágica se conecta con la muerte de las células neuronales, lo que explica la neurodegeneración asociada con la LSD. Además, la ALP se induce en condiciones de inanición, es decir, durante la baja disponibilidad de energía o nutrientes para reponer los nutrientes. La ALP es esencial para la mitofagia y, por lo tanto, para la función mitocondrial adecuada, lo que destaca aún más el vínculo estrecho entre la ALP y los recursos energéticos. Además, la falla en la proteostasis y la agregación de proteínas provoca el secuestro de proteínas, lo que impide sus actividades fisiológicas en procesos como la dinámica del citoesqueleto, la función sináptica y la homeostasis energética. Además, la ALP está estrechamente relacionada con la (dis)función sináptica y axonal. Por lo tanto, colectivamente, la proteostasis interrumpida puede interactuar negativamente con múltiples características del NDD, pintando una imagen de cómo varias características pueden funcionar juntas para inducir la neurodegeneración (Figura 3) .
Anormalidades del citoesqueleto
El citoesqueleto neuronal consta de tres estructuras poliméricas principales que interactúan entre sí, que se distinguen por su composición proteica y su diámetro: (1) microtúbulos basados en tubulina, (2) filamentos intermedios (neurofilamentos) y (3) microfilamentos basados en actina . Estas estructuras permiten que las neuronas construyan, mantengan y transformen su arquitectura, así como también facilitan la organización y el transporte de cargas intracelulares y mitocondrias a lo largo de sus longitudes extendidas, apoyando así la homeostasis energética y la función sináptica. Las estructuras presinápticas y postsinápticas tienen un citoesqueleto especializado y dinámico para soportar su función dinámica, estructura y altas demandas de energía, sirviendo como sitios de anclaje para las mitocondrias (microtúbulos) y como impulsores de cambios plásticidad (actina). El transporte axonal regula el transporte de proteínas, lípidos, mRNA y orgánulos, incluidas las mitocondrias, a las sinapsis, lo que ejerce un papel crucial en la neurotransmisión, la señalización trófica y las respuestas al estrés. Los NDD están asociados con alteraciones del citoesqueleto neuronal que conducen a la pérdida del capacidad de transmitir información y carga, incluidas las mitocondrias para satisfacer las demandas de energía y los componentes centrales esenciales entre el cuerpo celular y las terminaciones sinápticas, lo que a menudo resulta en un proceso de muerte hacia atrás (o hacia adelante) (Figuras 1 y 3).
El descubrimiento de mutaciones en el gene del filamento de luz neuronal intermedio (NEFL) en la enfermedad de Charcot-Marie-Tooth (CMT) y MAPT13 que codifican la proteína de unión a microtúbulos tau en las tauopatías (p. ej., FTD-tau, CBD y PSP13) es evidencia directa por la importancia de la función citoesquelética alterada en los NDD. Además, muchos NDD contienen agregados de proteínas del citoesqueleto neuronal, es decir, tau, actina o neurofilamento como ALS y SMA, entre otros. Además, el fenotipo neurodegenerativo en varios modelos de ratón se ha relacionado con mutaciones espontáneas en componentes de la maquinaria del citoesqueleto. Estos datos indican que los defectos en el citoesqueleto neuronal y funciones asociadas, como el transporte axonal, juegan un papel central en NDD. El papel de la lesión axonal en los NDD también se refleja en la extensa neurodegeneración en los NDD, donde los axones distales se separan del cuerpo celular, lo que resulta en la acumulación de materiales al final del muñón axonal y la ruptura del citoesqueleto axonal. En SCI, la lesión axonal también se asocia con isquemia, agotamiento de energía, excitotoxicidad e inflamación, lo que impulsa aún más el proceso neurodegenerativo. función y supervivencia de las células neuronales.
Se reconoce que la disfunción y la degeneración axonal contribuyen de forma destacada a la progresión de la enfermedad en la ELA, la MS, la TBI y el PD, así como en el SNP y los trastornos oculares, trastornos espinocerebelosos y neuropatías periféricas, la destrucción de las regiones distales de los axones largos precede a la degeneración manifiesta de los cuerpos celulares neuronales durante meses o años, lo que eventualmente conduce a la degeneración del soma a través de un proceso de degeneración retrógrada o patología de muerte regresiva. En la AD y las tauopatías , la hiperfosforilación de tau y/o la desregulación de tau y la relación 3R/4R de tau afectan la unión de tau a los microtúbulos que, a su vez, se asocia con una alteración de la dinámica de los microtúbulos axonales y del transporte axonal. Cualquiera que sea el mecanismo y la extensión del citoesqueleto colapso en estos NDD, este evento celular probablemente participa en el proceso de neurodegeneración a través de pasos moleculares que implican la pérdida de un el transporte axonal y la consiguiente distribución subcelular inadecuada de vesículas y orgánelos clave como las mitocondrias, que afectan indirectamente al metabolismo energético y la función sináptica, aunque también deben tenerse en cuenta las anomalías del citoesqueleto en la sinapsis.
En particular, los agregados de neurofilamentos se detectan en varios NDD. Los agregados de neurofilamentos forman una red de gel de cristal líquido en ALS, AD, PD, FTD, SMA, SCA1 y otros NDD. Aunque el mecanismo subyacente a la agregación de proteínas en estas situaciones sigue siendo en gran parte desconocido, la hiperfosforilación de los filamentos intermedios parece estar implicada. Además, la agregación de actina, con un papel clave en la dinámica sináptica, ocurre en varios NDD, y probablemente contribuye a los procesos neurodegenerativos. En la última década, las concentraciones de neurofilamentos en biofluidos han surgido como un biomarcador clínico prometedor para la neurodegeneración en muchos trastornos neurológicos, incluidos ELA, MS, TBI, accidente cerebrovascular y demencias, lo que subraya aún más que los defectos del citoesqueleto son un sello común de los NDD. Tras la degeneración del axón afectado, se cree que los neurofilamentos se liberan en el LCR y llegan a la sangre periférica, donde pueden medirse en concentraciones femtomolares.
Finalmente, la alteración del citoesqueleto puede interactuar con otras características del NDD, en particular la pérdida del mantenimiento sináptico y el metabolismo energético alterado, el transporte de RNA, la agregación de proteínas y la autofagia, y la muerte neuronal (Figura 3). La liberación de proteínas del citoesqueleto (incluidos neurofilamentos y tau) puede inducir reacciones inflamatorias, que pueden agravar aún más los defectos del citoesqueleto existentes.
Homeostasis energética alterada
Como las neuronas son células del cuerpo humano altamente activas y energéticamente exigentes, se ha demostrado que los defectos en el metabolismo energético participan en muchos NDD diferentes (Figuras 1 y 3). El ATP es la molécula clave del metabolismo energético del cerebro, que puede ser alimentado por el metabolismo de la glucosa,cetonas o el lactato, y se genera mediante la fosforilación oxidativa en las mitocondrias a través de la cadena de transporte de electrones. Los sustratos energéticos (glucosa/lactato) pueden suministrarse directamente desde el torrente sanguíneo. a las neuronas o indirectamente a través de los astrocitos. Un deterioro directo o indirecto en la función mitocondrial está involucrado en el proceso patogénico de varios NDD, probablemente debido a la baja disponibilidad de ATP y la consiguiente alteración de la funcionalidad de los procesos que demandan mucha energía en las neuronas, particularmente en las sinapsis, como el equilibrio iónico (bombas de membrana que consumen ATP), la homeostasis del calcio, la dinámica del citoesqueleto y la proteostasis. Además, la disfunción mitocondrial puede provocar estrés oxidativo como resultado de una mayor liberación de electrones libres que reaccionan con el oxígeno o el nitrógeno, dando lugar a daño macromolecular vía ataque de especies reactivas de oxígeno (ROS) de proteínas, lípidos y/o ácidos nucleicos. Tales alteraciones intracelulares pueden estimular la disfunción neuronal y la eventual muerte celular (Figura 3).
El papel de la homeostasis energética en la neurodegeneración se refleja en el accidente cerebrovascular, donde la excitotoxicidad y el agotamiento de la energía impulsan conjuntamente los procesos neurodegenerativos. De manera similar, los efectos concomitantes de la lesión axonal, la excitotoxicidad y el agotamiento de la energía promueven la neurodegeneración en la SCI. Evaluación de la homeostasis energética se puede realizar utilizando FDG-PET, que permite medir el metabolismo regional de la glucosa en el cerebro. La homeostasis energética alterada se refleja en la FDG-PET alterada en la mayoría, si no en todos, los NDD. Además, muchas enfermedades causadas por defectos hereditarios en las enzimas de la glucólisis, el metabolismo de los lípidos o el metabolismo mitocondrial presentan síntomas de mal funcionamiento del sistema nervioso. Varios trastornos raros que muestran defectos neurológicos también surgen de mutaciones en el DNA mitocondrial (DNAmt) que alteran la funcionalidad de los complejos de proteínas de la cadena respiratoria, lo que lleva a una mala utilización del oxígeno, estrés oxidativo y reducción de la producción de ATP.
Los síndromes mitocondriales clásicos que se derivan de mutaciones patogénicas en el genoma nuclear, que codifica proteínas mitocondriales que desempeñan funciones críticas en una variedad de procesos, incluido el mantenimiento del DNAmt, que se presenta con diversos síntomas, incluidas características neurológicas. Estas observaciones clínicas enfatizan la importancia de la integridad y función mitocondrial adecuadas para respaldar la salud de los órganos y tejidos. , particularmente aquellos con altas demandas de energía como el cerebro. Además, se ha informado que varios genes relacionados con NDD, en particular PD (Parkin, PINK1) y CMT (mitofusin2), regulan las vías de control de calidad mitocondrial.
La disfunción mitocondrial también ocurre en varios NDD con etiología no mitocondrial. Por ejemplo, estudios de modelos preclínicos, tejidos de pacientes y pacientes con NDD indican que la disfunción mitocondrial es parte del proceso patogénico en AD, HD y ALS, proteínas que incluyen α-sinucleína y tau modulan la fisión mitocondrial dependiente de actina. Como se mencionó en la sección anterior, las mutaciones y las proteínas agregadas que afectan el transporte axonal y la dinámica del citoesqueleto pueden afectar negativamente el transporte mitocondrial. Aunque en la mayoría de los casos se desconoce el origen de la disfunción mitocondrial, estos ejemplos indican que existen amplios problemas mitocondriales en los NDD que afectan negativamente la función mitocondrial, el control de calidad o el transporte.
Las consecuencias del daño mitocondrial o la desregulación de la célula pueden ser amplias (Figura 3). Por ejemplo, el daño mitocondrial puede conducir a alteraciones de la homeostasis del Ca2+, lo que da como resultado niveles elevados de Ca2+ intracelular y actividades enzimáticas dependientes de Ca2+ desreguladas, liberación de enzimas lisosomales y degradación del citoesqueleto, proteínas, lípidos y DNA. La reducción de la función mitocondrial y la síntesis de ATP también se asocia con una producción más baja del eliminador de glutatión (GSH), lo que podría empeorar el entorno oxidativo de la célula. La función mitocondrial deteriorada y el suministro de energía más bajo a las neuronas también afectan funciones fisiológicas clave, como la homeostasis iónica a través de la regulación de los canales iónicos; homeostasis del calcio, que afecta a las enzimas de degradación; dinámica del citoesqueleto; y rutas de producción y degradación de proteínas y orgánulos. Por lo tanto, la homeostasis energética alterada está estrechamente relacionada con varias características, incluida la muerte de las células neuronales, como se analiza a continuación.
Defectos de DNA y RNA
A la acumulación de daños en el DNA y los defectos en el metabolismo del RNA se les ha asignado un papel fundamental en una amplia gama de NDD (Figuras 1 y 3), su deterioro y daño por una amplia gama de agentes intracelulares o ambientales.
En el SNC, se supone que las principales genotoxinas son las ROS generadas como subproductos de la fosforilación oxidativa mitocondrial. Las alteraciones persistentes en el DNA pueden provocar eventos moleculares adversos, como mutagénesis, reordenamientos cromosómicos, detención de la transcripción del RNA o colapso de la horquilla de replicación del DNA, eventos que pueden promover la disfunción celular y la muerte celular. Para evitar estos puntos finales patogénicos, se han desarrollado respuestas complejas y sistemas de reparación para preservar la integridad del DNA y garantizar la funcionalidad normal del genoma. De manera similar, existen mecanismos intrincados que generan, utilizan y procesan fielmente la molécula de RNA para asegurar operaciones celulares apropiadas. El metabolismo y la homeostasis del RNA, que abarcan procesos como la transcripción, el empalme, el transporte y la degradación del RNA, la traducción y la biogénesis de los RNA reguladores no codificantes, es un conjunto complejo que implica numerosas interacciones con las proteínas de unión al RNA y las especies de RNA. Anomalías en cualquiera de los componentes de la regulación del RNA tiene consecuencias sobre la traducción de proteínas, la agregación de proteínas y la interferencia de RNA (RNAi). En cuanto a la formación de gránulos de estrés característicos (SG) que involucran ribonucleoproteínas (RNP). Además, la traducción RNA (traducción no ATG codificada por RNA asociada a la repetición) puede conducir a la generación de diferentes proteínas repetidas.
Defectos de DNA
La participación del daño del DNA en la neurodegeneración se destaca por el hecho de que varios trastornos hereditarios raros que presentan complicaciones neurológicas son el resultado de defectos en la capacidad para responder de manera eficiente y eliminar el estrés genómico. Por ejemplo, mutaciones en genes que codifican proteínas que normalmente funcionan para resolver las roturas de doble cadena del DNA (DSB) o el estrés replicativo (p. ej., ataxia telangiectasia [AT]) pueden conducir a la atrofia cerebral más adelante en la vida, en consonancia con el daño del DNA endógeno que promueve la pérdida progresiva de células neuronales. Varios NDD, en su mayoría ataxias autosómico recesivas, también se originan a partir de defectos hereditarios en la capacidad de resolver roturas de cadena sencilla (SSB) del DNA, productos frecuentes del ataque de ROS al DNA, de las vías de muerte celular en neuronas que no se dividen, mientras que el daño SSB se elimina fielmente mediante la reparación de recombinación homóloga dirigida por replicación en células en división. Alternativamente, el daño persistente en el DNA promueve la activación del ciclo celular y la reentrada, lo que lleva a la muerte celular apoptótica de las neuronas posmitóticas. En consonancia con el papel prominente de las lesiones que bloquean la transcripción que conducen a la enfermedad neurológica, los defectos heredados en los componentes de la subunidad acoplada a la transcripción vía de reparación por escisión de nucleótidos (TC-NER) dan lugar a la neurodegeneración. Las mutaciones recesivas en senataxina (SETX), una helicasa de RNA-DNA, están vinculadas a la ataxia con apraxia oculomotora tipo 2 (AOA2), mientras que las mutaciones dominantes raras se observan en una forma de ELA de inicio juvenil que implica bucles R genómicos (es decir, híbridos de RNA-DNA, intermediarios frecuentes de la transcripción) en la muerte de las células neuronales.
Fuera de los trastornos de reparación del DNA señalados anteriormente, los defectos en el procesamiento del daño del DNA se han relacionado con muchos otros NDD, incluidos casos esporádicos y familiares de AD, PD, HD y ALS. . La evidencia sugiere que la reparación comprometida del daño oxidativo del DNA puede ser un modificador del riesgo de degeneración neurológica en modelos de AD y patología similar a l PD. ,debido a la expresión reducida de proteínas clave de respuesta DSB. Además, las proteínas asociadas con tauopatías (tau), ALS/FTD (TDP-43 y FUS), HD (Htt), ataxia espinocerebelosa (ATXN2) o SMA (SMN1/SNM2) participan en varias respuestas al daño del DNA, más comúnmente en el contexto de roturas de cadenas de DNA. Curiosamente, los agregados de α-sinucleína pueden activar la quinasa de respuesta DSB mutada en AT (es decir, ATM), mientras que los niveles elevados de poli (ADP) ribosa, un producto de la hiperactividad de la poli(ADP-ribosa) polimerasa 1 (PARP1), acelera la fibrilación de la α-sinucleína. La evidencia reciente también indica que la acumulación de daño en el DNA nuclear conduce a la hiperactivación del DNA sensor de imagen PARP1, y el consecuente consumo de NAD y disfunción mitocondrial.
Defectos de RNA
Los datos acumulados indican un papel importante para la desregulación del RNA en varios NDD, al alterar las funciones fisiológicas o al inducir toxicidad del RNA o la formación de SG que involucran la agregación de proteínas. Por ejemplo, los defectos de RNA juegan un papel en la etiología de la enfermedad de ALS/FTD y enfermedades polyQ. , y en el transporte de RNA. De manera similar, las mutaciones patógenas en FUS causan la mala localización de la proteína normalmente nuclear en el citoplasma, lo que lleva a defectos en el metabolismo del RNA, específicamente en las transcripciones que codifican proteínas que regulan el crecimiento dendrítico y las funciones sinápticas. Los defectos del metabolismo del RNA también están relacionados con FTD/ALS vinculado a C9orf. En las enfermedades poliQ, tanto las expansiones de CAG, que pueden provocar un secuestro anómalo de proteínas, como la traducción de RNA, que conduce a la generación de proteínas repetidas, se consideran elementos génicos a través del atrapamiento de proteínas reguladoras clave. Ejemplos adicionales que respaldan los defectos del RNA como mecanismo impulsor de los NDD incluyen la proteinopatía multisistémica (MSP), causada por mutaciones en el gene nuclear heterogéneo RNP A1 (HNRNPA1) o A2B1 (HNRNPA2B1), y SMA, ya que SMN1/SMN2 son factores de transacción involucrados en el empalme .
También se cree que la formación de agregados citoplasmáticos de SG desempeña un papel importante en el proceso de NDD. Los SG son complejos RNP citosólicos densos que se forman en respuesta al estrés celular para inhibir temporalmente la traducción y almacenar mRNA. Varias proteínas de unión a RNA asociadas con FTD y ALS (es decir, TDP-43, FUS, EWSR1, TAF15, hnRNPA1, hnRNPA2B1, ATXN2, TIA1 y VCP) están involucradas en la dinámica de SG. Además, los DPR que contienen arginina pueden alterar la composición y dinámica de SG. La formación de SG puede ser inducida por UPS disfuncional, que culmina en la acumulación de proteínas ubiquitinadas. Por lo tanto, los cambios en el metabolismo del RNA se asocian con proteostasis alterada, otro sello distintivo de los NDD.
Algunos NDD que implican la expansión de repeticiones de microsatélites pueden originarse en parte por el secuestro de proteínas de unión a RNA. que contiene RNA que forma agregados y focos de RNA, secuestrando proteínas de unión al RNA con funciones críticas en el empalme alternativo. Se observa un fenómeno similar en los trastornos asociados a la repetición, el síndrome de temblor/ataxia asociado al cromosoma X frágil (FXTAS) y ALS/FTD. Colectivamente, la homeostasis alterada del RNA está relacionada con la producción alterada de proteínas y la agregación de proteínas, y la formación de SG conduce al secuestro de proteínas que afecta la funcionalidad, incluidas las proteínas que mantienen la función sináptica. Con los vínculos ya presentados entre el metabolismo/homeostasis del RNA y otras características del NDD (p. ej., transporte axonal de mRNA, generación local de proteínas en las sinapsis, homeostasis energética y proteostasis) (Figura 3), es evidente que estos procesos moleculares pueden afectarse entre sí.
Inflamación
La neuroinflamación, incluidas la microgliosis y la astrogliosis, es un sello patológico de los NDD, incluidos la AD, la PD, la ELA, la HD y el accidente cerebrovascular (Figuras 1 y 3). Además de la presencia invariable de inflamación en muestras de cerebro post mortem de pacientes con NDD, se demuestra un papel definitivo de la neuroinflamación en la neurodegeneración en la enfermedad neuroinflamatoria prototípica, la MS y los NDD relacionados.
La microgliosis se detecta invariablemente en todos los NDD, incluidas las proteinopatías y las no proteinopatías. La microglía normalmente ejerce funciones de sensor, limpieza y defensa en el cerebro, y las aberraciones en estas funciones pueden conducir a la neurodegeneración. Al defender la función cerebral, la microglía suele reaccionar ante patógenos o signos de lesión. Tras la activación, la microglía está involucrada en la producción de citocinas/quimiocinas, la activación de la fagocitosis, la desregulación de las funciones fisiológicas y la producción de ROS. Después de un factor estresante agudo, el proceso inflamatorio normalmente se resuelve activamente. Sin embargo, la falta de resolución da como resultado una inflamación crónica que puede promover el proceso neurodegenerativo. La activación no resuelta de la microglía puede ocurrir en los NDD debido a la presencia de señales de peligro, como agregados de proteínas, proteínas mal plegadas, sinapsis dañadas, entrada de Ca2+ o ROS mitocondriales , es decir, mecanismos que impulsan otros sellos distintivos de NDD. Varias proteínas NDD agregadas inducen la activación microglial, como Aβ, tau, α-sinucleína, fibrillas PrP o SOD1. Además, se cree que ocurre una transición entre diferentes estados microgliales durante el proceso de la enfermedad, con poblaciones particulares asociadas específicamente con la neurodegeneración ( por ejemplo, microglía asociada a la enfermedad [DAM] o fenotipo neurodegenerativo microglial [MgND]). Estas poblaciones microgliales provocan funciones únicas en la regulación de la producción de citocinas, la fagocitosis, la producción de ROS o las interacciones astrogliales, lo que finalmente afecta la función sináptica y la muerte de las células neuronales. Los conocimientos sobre las funciones perjudiciales y protectoras de las diferentes poblaciones microgliales serán importantes para una orientación terapéutica eficaz en los NDD.
Los estudios genéticos también respaldan un papel activo de la microglía en los NDD. Por ejemplo, la identificación de mutaciones con pérdida de función del receptor desencadenante expresado en las células mieloides 2 (TREM2) en individuos con la enfermedad de Nasu-Hakola, un trastorno raro asociado con la neurodegeneración que conduce a la demencia y a la muerte prematura, reveló un papel crucial para la microglía. Como TREM2 solo se expresa en la microglía del cerebro, estas células deben contribuir a la degeneración del SNC en este trastorno. Las variantes genéticas de TREM2 también están asociadas con el riesgo de AD en el estado heterocigoto, y TREM2, junto con la apolipoproteína E (ApoE ), juega un papel crucial en la transición de microglía homeostática a microglía asociada con fenotipos de enfermedades. De hecho, tanto ApoE como TREM2 modifican la agregación de proteínas y la neurodegeneración en modelos preclínicos de NDD. La función de TREM2 parece proteger contra la toxicidad local relacionada con Aβ, mientras que más adelante en la fase de tauopatía de la enfermedad, su función puede ser perjudicial. La ApoE, que actúa a través de la microglía, desempeña un papel importante en la conducción de la neurodegeneración mediada por tau, la expresión de GRN aumenta la microgliosis y la poda sináptica, lo que lleva a la hiperexcitabilidad. Curiosamente, Htt(hungtintina) también se expresa fuertemente en la microglia, y la expresión de Htt mutante en la microglia promueve la activación microglial autónoma y se asocia con una mayor neurodegeneración.
Los estudios de asociación del genoma completo en la AD han identificado muchos genes importantes para la función de la microglía, la endocitosis y el metabolismo de los lípidos. Está emergiendo una estrecha relación entre el metabolismo de los lípidos y la activación de la microglía a medida que los genes relacionados con los lípidos aumentan en la transición asociada. de la microglia asociada del estado homeostático al daño. Además, existe evidencia genética de contribuciones microgliales a sinucleinopatías esporádicas. Al igual que en AD, la ApoE actúa como un factor de riesgo para la demencia en el contexto de la enfermedad con cuerpos de Lewy, y hay evidencia de que la ApoE está involucrada en la neurodegeneración de la sinucleinopatía. GRN también es un factor de riesgo genético compartido para múltiples NDD, incluida la AD. Finalmente, LRRK2 se expresa en microglia, donde se ha propuesto modificar el riesgo de PD esporádica. Como consecuencia, la microglia podría contribuir a la patogenia de múltiples NDD. El hecho de que esta asociación sea causal de la neurodegeneración y no simplemente un efecto espectador está respaldado por estudios en modelos preclínicos, por ejemplo, en ratones transgénicos SOD1G37R, donde se observó que el proceso neurodegenerativo se retrasa considerablemente al eliminar la SOD1 mutante de los macrófagos y la microglía , la eliminación microglial a través de la inhibición de CSF1R modifica la progresión en modelos de AD, tauopatías,PrD y TDP-43 ALS.
En particular, la microglía interactúa estrechamente con los astrocitos y los activa, que también son críticos para el mantenimiento de la salud y la función neuronal. Ha surgido un papel activo de los astrocitos en el proceso neurodegenerativo de varios NDD ). Brevemente, los astrocitos son cruciales para el funcionamiento neuronal adecuado y ejercen un papel clave en la preservación de la homeostasis del glutamato y son una parte vital de la red tri-sináptica. Los astrocitos liberan factores extracelulares, incluidas las quimiocinas y las citocinas, y desempeñan un papel clave en la cicatrización glial. . Al modular estos diferentes procesos, los astrocitos pueden contribuir directamente a los defectos sinápticos y la neurodegeneración de una manera no autónoma de las células (Figura 3). Es importante destacar que los astrocitos normalmente se activan en la proximidad de las neuronas dañadas, lo que lleva a la gliosis reactiva. Los astrocitos también pueden ser un objetivo directo de la patología de agregación de proteínas en algunos NDD. También existe una estrecha red de señalización recíproca entre los astrocitos y la microglia. Se han utilizado biomarcadores de fluidos y estudios de imágenes PET para detectar y monitorear las contribuciones gliales a los NDD, incluidas aplicaciones que involucran ligandos PET específicos de microglia y LCR o biomarcadores basados en sangre como proteína ácida fibrilar glial (GFAP), TREM2 y ciertas citoquinas. Estos estudios han revelado vínculos estrechos, específicos de la etapa de la enfermedad, entre la activación glial y el proceso NDD.
Finalmente, la inflamación, es decir, los astrocitos reactivos y la microglía reactiva, activados por las diferentes características del NDD e interactuando con ellas, pueden funcionar en combinación con otras características del NDD para acelerar el proceso de la enfermedad (Figura 3). Como se describió anteriormente, la activación de los astrocitos afecta la función sináptica, la homeostasis energética, la agregación de proteínas y la neurodegeneración, mientras modula recíprocamente la microglía. Por el contrario, la microglía hiperactiva desempeña un papel en la eliminación de sinapsis en los NDD, lo que podría contribuir a la disfunción de la red neuronal. Como se describió anteriormente, la activación de la microglia por la agregación de proteínas y el papel de la microglía en afectar la agregación de proteínas están bien establecidos, lo que indica una interacción adversa entre estos dos fenómenos. En respuesta a las señales de peligro, la microglía secreta ROS, lo que se suma al daño oxidativo y altera la homeostasis energética en el cerebro. La microglía elimina las neuronas que expresan señales “cómeme” en la superficie celular. Estos ejemplos resaltan la estrecha interconexión entre la inflamación y otras características del NDD.
Muerte de células neuronales
Varias propiedades inherentes de las neuronas pueden hacerlas especialmente vulnerables a la muerte celular en NDD (Figuras 1 y 3). Estos incluyen: (1) su naturaleza posmitótica que da como resultado (a) la acumulación gradual de daño asociado con la edad en el DNA, los lípidos, las proteínas y los orgánelos y (b) la incapacidad para replicarse y reponer la población de células neurales; (2) sus altos requerimientos de energía, principalmente debido a la necesidad de apoyar la función sináptica y la producción de ROS asociada a través de la fosforilación oxidativa mitocondrial; (3) sus axones y dendritas extendidos, lo que lleva a un requisito de transporte y organización estructural a largas distancias; y (4) su dependencia de las células gliales para su mantenimiento, energía y defensa. Como resultado, las neuronas parecen ser más susceptibles a la muerte celular, una característica que se ve exacerbada por una disminución de los mecanismos de resiliencia asociada con la edad. Las características de NDD descritas anteriormente contribuyen individual y colectivamente a la pérdida de células neuronales, presumiblemente de una manera que da lugar a distintas manifestaciones patológicas y clínicas. Se ha propuesto que diferentes características del NDD actúen en conjunto para anular en última instancia la resiliencia neuronal intrínseca a las agresiones internas y externas (Figura 3).
La muerte neuronal, que en última instancia resulta en la pérdida de volumen cerebral que puede monitorearse mediante resonancia magnética volumétrica y en la liberación de proteínas intraneuronales como tau y neurofilamentos en biofluidos, se presenta en diferentes formas. Se puede hacer una clasificación específica basada en el mecanismo de la muerte o el inductor de la muerte. Los mecanismos de muerte neuronal mejor descritos incluyen la apoptosis y la necrosis intrínsecas y extrínsecas, pero otros incluyen la necroptosis, la ferroptosis, la fagoptosis y la muerte celular autofágica , la piroptosis y la transición de la permeabilidad mitocondrial también están bien documentadas. Aunque existen diferentes tipos de muerte neuronal, existe una diafonía entre los mecanismos, y es posible que si una neurona no ejecuta un programa específico de muerte celular, otra lo tome. Muchos de los desencadenantes inmediatos de la muerte de las células neuronales ya se han mencionado e incluyen: axotomía, reingreso aberrante del ciclo celular, excitotoxicidad y oxitosis del glutamato, pérdida de neuronas conectadas, proteínas agregadas, respuesta de proteína desplegada (UPR), ruptura lisosomal, autofágia, oxidantes y daño macromolecular (DNA). Por lo tanto, múltiples características del NDD discutidas en esta revisión son inductores de la muerte de las células neuronales y se espera que impulsen sinérgicamente el proceso neurodegenerativo (Figura 3).
La comprensión de los actores moleculares y las vías involucradas en la muerte celular es importante para comprender los mecanismos de la neurodegeneración. La excitotoxicidad provoca la muerte neuronal por exceso de Ca2+ citoplasmático, activando varios programas de muerte neuronal mediante la apertura del poro de transición de permeabilidad mitocondrial (mPTP) y activando las calpaínas que promueven la apoptosis o la necrosis por muerte de las células lisosomales.
El agotamiento de la energía provoca la muerte de las células neuronales por un mecanismo diferente , donde la privación de oxígeno y glucosa da como resultado un rápido agotamiento de ATP en las neuronas que conduce a la despolarización de la membrana plasmática y la subsiguiente activación de los canales de Ca2+ dependientes de voltaje presinápticos y somatodendríticos y la liberación de glutamato.
Las vías de muerte celular también pueden iniciarse directamente por el daño mitocondrial y la disfunción mitocondrial asociada. Por ejemplo, tras la alteración de la integridad mitocondrial, el factor inductor de la apoptosis (AIF) puede trasladarse de la mitocondria al núcleo e inducir la condensación de cromatina independiente de caspasa y la escisión del DNA. Además, tras la permeabilización de la membrana mitocondrial externa, el citocromo c (CytC ) puede liberarse, lo que da como resultado la generación de un complejo de apoptosoma que contiene CytC, caspasa-9 y apaf-13. Este proceso asociado a las mitocondrias puede ser inducido por varios estresores celulares, incluidos el daño del DNA, el estrés oxidativo, los glucocorticoides, la producción de ceramida o la pérdida de factores de crecimiento . La ruptura lisosomal conduce a la pérdida de células neuronales por la liberación de enzimas lisosomales, incluidas las catepsinas y las hidrolasas.
Además de las vías de muerte celular autónoma, se ha demostrado que los procesos autónomos no celulares contribuyen a la pérdida neuronal. Por ejemplo, las células apoptóticas pueden ser fagocitadas por la microglía activada, lo que limita la neuroinflamación en curso (Figura 3). Las neuronas apoptóticas se etiquetan para la fagocitosis al exponer la fosfatidilserina (PS) como una señal de “comerme”. En condiciones fisiológicas normales, la PS no es accesible, aunque durante la apoptosis, la división de los transportadores de PS dependiente de caspasa da como resultado la exposición de la superficie celular de la PS. Curiosamente, recientemente se demostró que la exposición a la PS ocurre en neuronas que muestran formas patológicas de tau. Por lo tanto, la agregación de proteínas y las vías de muerte celular autónomas no celulares interactúan. Sin duda, las características de los NDD convergen en la muerte neuronal, ya que la etapa final de los NDD es la pérdida de la población de células neuronales que conduce a una funcionalidad deteriorada.
Características de los NDD: un marco para un enfoque holístico para estudiar los NDD
A continuación, se proporciona un marco para un enfoque holístico para estudiar los NDD y unificar y categorizar diferentes NDD en función de sus características principales de NDD. Proporcionamos este marco como una suma de los factores genéticos y las vías bioquímicas que contribuyen a los NDD, descubriendo procesos superpuestos entre los NDD. Considero específicamente ocho características comunes de los NDD: agregación patológica de proteínas, disfunción de la red neuronal y sináptica, proteostasis aberrante, anomalías del citoesqueleto, metabolismo energético alterado, defectos de DNA y RNA, inflamación y muerte de células neuronales.
La interconexión de los sellos distintivos de NDD destaca la necesidad de terapias multidirigidas
Una idea importante de esta discusión es que los diferentes sellos distintivos de NDD muestran un alto grado de interconexión (Figura 4). Por ejemplo, la disfunción sináptica y la excitotoxicidad están estrechamente relacionadas con la homeostasis energética, el estrés oxidativo y la muerte neuronal. También se han identificado relaciones directas entre agregación proteica patológica, proteostasis aberrante, función sináptica, excitotoxicidad y muerte neuronal. De manera similar, la homeostasis energética, el estrés oxidativo, el daño del DNA y la muerte neuronal son procesos altamente interactivos. Las vías autónomas no celulares que involucran a la glía son importantes en la modulación de la función neuronal, la eliminación de sinapsis, la neurodegeneración y la neuroinflamación. Estos ejemplos destacan los estrechos vínculos entre las características y la interconexión de las mismas, lo que sugiere que la resistencia neuronal a las agresiones puede verse anulada por los efectos adversos de eventos simultáneos y sinérgicos que promueven la muerte neuronal.
La interconexión de los NDD debe tenerse en cuenta en las estrategias terapéuticas, ya que tales interacciones implican que dirigirse a un único sello distintivo del NDD puede ser insuficiente para detener el proceso neurodegenerativo. Cuando se apunta solo a un sello distintivo de NDD, el proceso de la enfermedad puede ser superado por uno de los otros sellos distintivos, lo que sugiere la necesidad de terapias combinatorias de objetivos múltiples para detener de manera efectiva los NDD. Aunque se han identificado terapias prometedoras basadas en oligonucleótidos antisentido (ASO) que utilizan el silenciamiento genético directo de un gene defectuoso en casos de AME, las terapias para enfermedades esporádicas siguen siendo esquivas. Se propone que un enfoque en el futuro sería ajustar el tratamiento de cualquier paciente en función de las características más destacadas del individuo según lo determinen los paneles de biomarcadores multimodales, apuntando a múltiples sistemas de manera concomitante durante la terapia, como se analiza a continuación.
Un marco para identificar puntos en común y diversificación entre y dentro de los NDD
A pesar de las características similares de los NDD, su importancia relativa en las diferentes enfermedades difiere claramente, probablemente dictada por factores genéticos y ambientales, así como por poblaciones neuronales específicas, regiones cerebrales o tipos de células afectadas. En este sentido, será importante definir los contribuyentes primarios y principales de la vía de la enfermedad y señalar los efectos más secundarios para cada NDD específico (Figura 4A) e incluso dentro de las subcategorías de NDD (Figura 4B). Por ejemplo, la agregación patológica de proteínas y la proteostasis aberrante, los defectos sinápticos y la inflamación podrían considerarse los principales contribuyentes a la AD, mientras que la proteostasis aberrante combinada con los defectos mitocondriales y las alteraciones en el metabolismo energético pueden desempeñar funciones importantes en la PD (Figura 4A). Las anomalías del citoesqueleto que se manifiestan en alteraciones axonales parecen ser primarias en las neuropatías periféricas. Por el contrario, la inflamación es un contribuyente claro y principal en las enfermedades inflamatorias prototípicas, como la MS. En la ELA, una combinación de defectos en la agregación de proteínas y en el metabolismo del RNA o del DNA, junto con defectos en las redes sinápticas y neuronales, parece estar crucialmente involucrada. Las PrD son ejemplos clásicos de enfermedades en las que la agregación y la propagación de proteínas son procesos patogénicos clave. Sin embargo, la disfunción neuronal y la muerte celular probablemente sean inducidas por una combinación de procesos NDD que impulsan sinérgicamente la neurodegeneración. Además de las diferencias obvias entre los NDD, sus puntos en común pueden sugerir que algunas estrategias podrían tener aplicaciones más amplias en todo el espectro de NDD y podrían ser útiles para enfocarse en múltiples características de NDD compartidas. Sin embargo, es necesario considerar los aspectos específicos de la enfermedad.
Un marco para la estratificación de subtipos dentro de NDD específicos para ensayos clínicos
Es importante destacar que el marco propuesto proporciona una base para la estratificación no solo entre NDD, sino también de subtipos dentro de NDD específicos (Figura 4B). Claramente existen diferencias individuales/personalizadas en los impulsores primarios dentro de los NDD específicos. Estas diferencias están definidas por factores genéticos, reflejados en la puntuación de riesgo poligénico, pero también por factores no genéticos que incluyen la edad, las prácticas de estilo de vida y las exposiciones ambientales. Las diferencias en la resiliencia y la vulnerabilidad neuronal de las diferentes regiones del cerebro en los NDD, pero también y en particular entre los individuos dentro de un NDD específico, determinarán las características distintivas primarias combinadas de NDD que impulsan el proceso de la enfermedad. La heterogeneidad entre los pacientes dentro de NDD específicos puede contribuir a los fracasos de los ensayos clínicos que se enfocan en un sello distintivo de NDD en particular, lo que apunta a la necesidad de estratificar a los pacientes. Es importante destacar que las patologías subyacentes a muchas de estas enfermedades comienzan muchos años antes de los síntomas clínicos. Para cuando surgen los síntomas, ya existe una pérdida neuronal y sináptica significativa. La identificación de individuos en las etapas presintomáticas de la enfermedad mediante el uso de biomarcadores será fundamental para los ensayos de prevención primaria y secundaria. Diferentes subtipos de NDD, según los perfiles moleculares, pueden presentarse con síntomas similares; sin embargo, los factores genéticos y externos pueden cambiar el peso y la contribución relativa de las diferentes características del NDD dentro de un NDD dado. Por ejemplo, dentro de la AD, es probable que existan pacientes que, además del fuerte componente de agregación de proteínas, muestren un fuerte componente inflamatorio, un fuerte componente sináptico o un fuerte componente de proteostasis/(ALP/UPS), con cada subtipo probablemente beneficiándose de manera diferente de un particular ( terapia multidirigida) (Figura 4B). Por lo tanto, las características de NDD pueden proporcionar un marco para la estratificación de pacientes dentro de NDD, definiendo subtipos para el diseño de terapias personalizadas con objetivos múltiples (Figura 4B).
En conjunto, se proporciona un marco para unificar y categorizar los NDD, así como para la estratificación de subtipos y pacientes dentro de NDD específicos, en función de las características de NDD identificadas por décadas de investigaciones genéticas y bioquímicas en modelos y pacientes. Se propone que la detención efectiva de los NDD requerirá biomarcadores mejorados para las características de NDD y terapias personalizadas multidirigidas, debido a la interconexión entre las características de NDD.
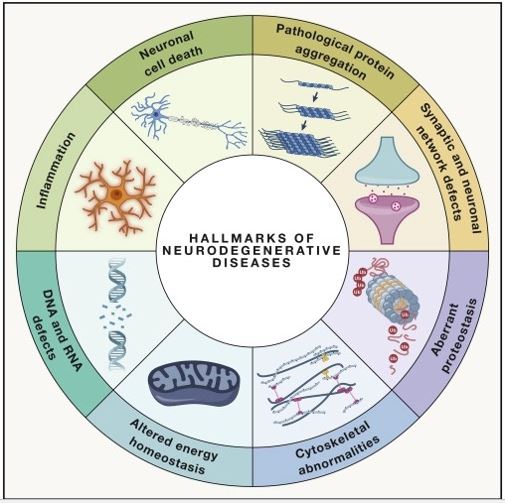
El esquema identifica e ilustra los ocho sellos distintivos descritos en el artículo. Con base en décadas de investigación básica, traslacional y clínica, se identificaron factores genéticos y vías bioquímicas subyacentes a muchos NDD, lo que resultó en la identificación de ocho características distintivas de NDD: agregación patológica de proteínas, disfunción de la red neuronal y sináptica, proteostasis aberrante, anomalías del citoesqueleto, alteración homeostasis energética, defectos de DNA y RNA, inflamación y muerte de células neuronales.
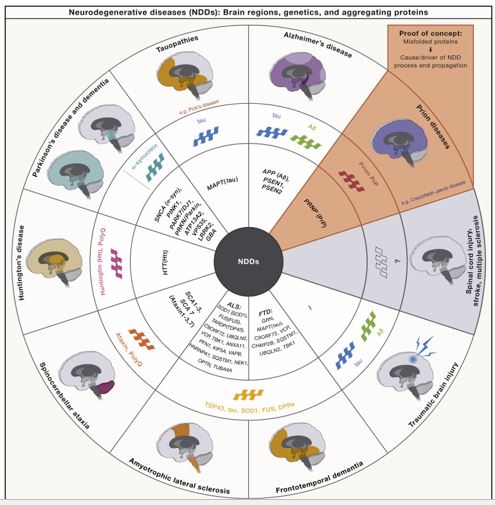
Los NDD respectivos y las regiones cerebrales afectadas se indican en el círculo exterior. Sus respectivos síntomas, las regiones cerebrales afectadas, los genes asociados y las proteínas de agregación se han descrito previamente en detalle. Las proteínas de agregación características en los NDD incluyen péptidos Aß formados por la escisión de la proteína precursora de amiloide (APP) (gene APP), tau (gene MAPT), α-sinucleína (gene SNCA), proteína 43 de unión al ADN TAR (TDP-43) (TARDBP gene ), superóxido dismutasa [Cu-Zn] (SOD1) (gene SOD1), proteínas de repetición dipeptídica (DPR) (gene C9orf72), proteína de unión a RNA FUS (gene FUS), huntingtina (PolyQ) (gene HTT), proteínas polyQ (PolyQ) y proteína priónica celular (PrPC) (gene PRNP). En el círculo interior (mayúsculas, cursiva) se indica una lista no exhaustiva de genes causales y de mayor riesgo vinculados a los diferentes NDD.
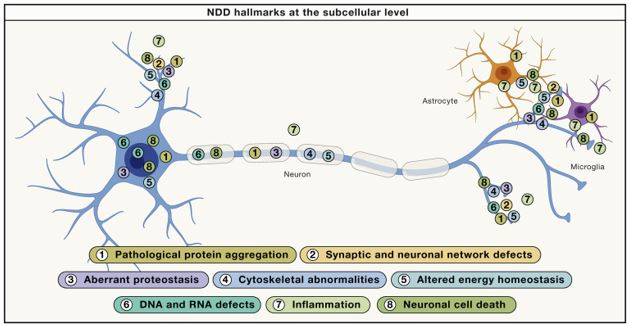
Una representación esquemática de las diferentes características de NDD (numeradas) y su ubicación en la neurona modelada y/o las células gliales acompañantes para la agregación patológica de proteínas (1), la disfunción de la red neuronal y sináptica (2), la proteostasis aberrante (3), el citoesqueleto anomalías (4), homeostasis energética alterada (5), defectos de ADN y ARN (6), inflamación (7) y muerte de células neuronales (8).
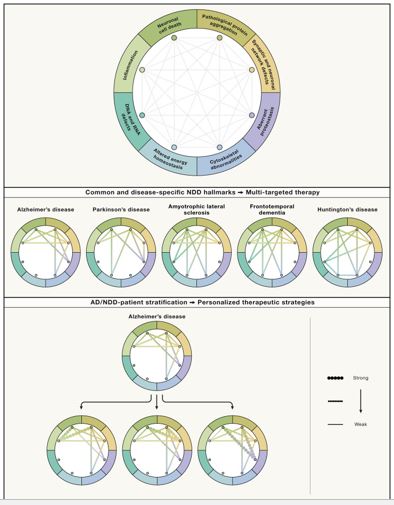
(A) La interconexión de los sellos se presenta esquemáticamente, proporcionando un marco para un enfoque holístico para estudiar los NDD. Este marco permite categorizar los NDD en función de sus características principales (A) se presentan ejemplos de NDD comunes, es decir, AD, PD, ALS, FTD y HD.
(B) Además, este marco permite identificar subtipos de pacientes dentro de NDD específicos, lo que permite la estratificación de los pacientes en función de sus principales características (B). Estos se definen por el insulto NDD y por la resiliencia neuronal y la vulnerabilidad del individuo. Se muestran ejemplos de subtipos dentro de AD. Por lo tanto, las estrategias de tratamiento de NDD se pueden diseñar en función de los perfiles distintivos individuales, lo que permite diseñar terapias personalizadas con objetivos múltiples.