La enfermedad renal crónica (ERC) es un problema de salud pública que afecta a más de 800 millones de personas en todo el mundo.1. La ERC puede ser causada por una variedad de procesos patológicos. Muchas causas son difíciles de identificar con el uso de diagnósticos clínicos tradicionales y, a menudo, las causas precisas siguen siendo desconocidas. A nivel mundial, la mayoría de los casos de ERC se han atribuido a diabetes mellitus o hipertensión arterial , que no se analizan aquí. Sin embargo, cada vez se reconocen más las causas genéticas de la ERC. La organización Kidney Disease: Improving Global Outcomes (KDIGO) destacó recientemente la importancia de la genética en la clasificación y el tratamiento de la ERC y recomendó a los médicos que consideren la posibilidad de realizar pruebas genéticas para mejorar la precisión diagnóstica y facilitar el tratamiento médico personalizado en nefrología.2.
Nuestra comprensión de la base genética de las enfermedades renales ha evolucionado considerablemente desde la identificación del locus de la enfermedad renal poliquística autosómica dominante (ERPAD) en 1985[3]. Desde entonces, se han identificado cientos de genes diferentes implicados en la enfermedad renal, muchos de ellos mediante secuenciación masiva paralela. Se han publicado en otros lugares listas de genes relacionados con la ERC4-6 que se actualizan continuamente.7.
Las enfermedades genéticas se han identificado con mucha más frecuencia entre los niños con ERC y solo recientemente han surgido como causas importantes de ERC en adultos[8-11]. Esta revisión analiza el diagnóstico y el tratamiento de la ERC de origen genético, centrándose en las formas monogénicas de la ERC y en las variantes que confieren un riesgo sustancial de ERC progresiva. Las variantes genéticas que son importantes solo en el contexto de las puntuaciones de riesgo poligénico[12] o los alelos de riesgo que modulan ciertas medidas de la función renal (por ejemplo, la tasa de filtración glomerular estimada o medida [TFGe o TFG][13] o la concentración de proteína urinaria) están más allá del alcance de esta revisión.
Epidemiología
Desde hace tiempo se sabe que muchas causas comunes y diversas de la enfermedad renal crónica se agrupan en familias, lo que indica contribuciones genéticas[14]. Además, las disparidades entre los grupos étnicos han sugerido contribuciones genéticas. La mayoría de las enfermedades renales genéticas son raras. Sin embargo, en conjunto, contribuyen sustancialmente a la prevalencia mundial de la enfermedad renal crónica. Múltiples informes mundiales indican que una variedad diversa de trastornos monogénicos distintos puede explicar aproximadamente entre el 30 y el 50 % de los casos de enfermedad renal crónica en niños[6,15–17], y aproximadamente entre el 10 y el 20 % de los casos en adultos[8–11,18]. Las enfermedades renales genéticas en la infancia y la adultez constituyen un continuo dependiente de la edad[11] sin un límite superior de edad para el diagnóstico[2].
=> Recibir por Whatsapp las noticias destacadas
Puntos clave
Genética de la enfermedad renal crónica
Las causas genéticas de la enfermedad renal crónica (ERC) no son poco comunes. Los pacientes con ERC deben ser derivados para una consulta y análisis genéticos cuando esté indicado.
Obtener un diagnóstico molecular a nivel de variante es importante, incluso cuando el diagnóstico basado en el fenotipo clínico respalda una causa genética específica. Los diagnósticos genéticos moleculares establecen causas precisas, con implicaciones clínicas para el seguimiento y el tratamiento personalizados y para un asesoramiento genético familiar eficaz.
Los diagnósticos monogénicos de enfermedad renal crónica más comunes en todo el mundo incluyen variantes patógenas en PKD y COL4A. Las variantes patógenas en muchos otros genes explican los diagnósticos genéticos restantes.
Algunos tipos de enfermedad renal crónica tienen determinantes genéticos complejos, y el riesgo de enfermedad depende del genotipo (p. ej., el genotipo APOL1) y del entorno. Las personas de ascendencia africana tienen más probabilidades que las personas de otras ascendencias de albergar variantes de riesgo de APOL1.
La investigación genética sobre la enfermedad renal crónica está creciendo rápidamente. Incluso los pacientes con resultados negativos de las pruebas genéticas deben ser reevaluados periódicamente a medida que se identifican nuevos genes y variantes de riesgo y las pruebas genéticas se vuelven más disponibles e informativas. Los diagnósticos genéticos de la enfermedad renal crónica pueden hacer que los pacientes sean elegibles para terapias genéticas existentes y, posiblemente, emergentes.
Mecanismos fisiopatológicos y clasificación
El riesgo genético de la enfermedad renal crónica también puede considerarse en un espectro de variantes genéticas de baja a alta penetración. En un extremo del espectro se encuentran las enfermedades mendelianas de alta penetración. El fenotipo patológico de estas entidades es causado por una variante patógena (o, en el caso de una enfermedad recesiva, dos variantes patógenas o una en estado homocigoto) en un solo gen. Estas afecciones suelen tener correlaciones estrechas entre genotipo y fenotipo. Por el contrario, las variantes de baja penetración o los alelos de riesgo suelen actuar en conjunto con factores ambientales para causar la enfermedad (Figura 1).
Las causas de la enfermedad renal crónica se pueden clasificar en función de las manifestaciones clínicas, las características histológicas renales o las características genéticas. Dada la magnitud de la heterogeneidad genética y fenotípica de la enfermedad renal crónica, las definiciones relacionadas con los genes son las más específicas y precisas. Un diagnóstico genético señala la causa raíz de una enfermedad determinada y puede potencialmente proporcionar información sobre los mecanismos moleculares y, a menudo, sobre el pronóstico y el tratamiento que van más allá de la información proporcionada por los fenotipos tradicionales de la ERC.
Las enfermedades renales genéticas se pueden agrupar en múltiples subcategorías que abarcan diversas malformaciones o síndromes atribuibles a condiciones específicas de un solo gen (Figura 2). Las definiciones relacionadas con los genes no necesariamente corresponden a manifestaciones clínicas específicas o características histológicas renales. Además, un diagnóstico genético conduce a la reclasificación del diagnóstico clínico o histológico original en el 10 al 50% de los casos[11,18,20]. Dicha reclasificación puede impulsar un cambio en el tratamiento hacia un enfoque centrado en los mecanismos genéticos y moleculares. Por lo tanto, las pruebas genéticas pueden complementar o incluso reemplazar a la biopsia renal como el estándar para el diagnóstico y el tratamiento de algunas formas de ERC. Estos conocimientos han dado como resultado una taxonomía molecular de la ERC que puede facilitar el uso de la medicina de precisión como práctica clínica estándar (Figura 2).
Genotipo y fenotipo en las formas genéticas de la enfermedad renal crónica
Hasta la fecha, cientos de genes se han asociado con las formas genéticas de la enfermedad renal crónica. Por lo tanto, resulta útil centrarse en los conocimientos relevantes para la práctica sobre las causas genéticas más comunes de la enfermedad renal crónica en adultos.
Enfermedades renales quísticas
Las enfermedades renales quísticas se deben con mayor frecuencia a ciliopatías renales, un grupo diverso de trastornos genéticos causados por alteraciones en las proteínas localizadas dentro del complejo cilio-centrosoma.21. Los fenotipos clínicos incluyen quistes renales múltiples, como se observa en la enfermedad renal poliquística autosómica dominante, y riñones ecogénicos de tamaño normal o pequeños, como se observa a menudo en la nefronoptisis.
PQRAD
La PQRAD es la enfermedad renal genética más común en todo el mundo y afecta entre el 4 y el 8 % de todos los pacientes con insuficiencia renal.22. La PQRAD es un trastorno sistémico con una expresión clínica variable que puede incluir manifestaciones extrarrenales como quistes hepáticos, aneurismas cerebrales y enfermedad valvular cardíaca.22. Las principales causas de la PQRAD son variantes patogénicas en dos genes, PKD1 (en aproximadamente el 78 % de los casos) o PKD2 (en aproximadamente el 15 % de los casos), que codifican las proteínas policistina 1 y policistina 2, respectivamente. Los estudios de correlación genotipo-fenotipo muestran que las variantes truncadas de PKD1 se asocian con una enfermedad renal más grave, en comparación con las variantes sin sentido de PKD1 y PKD2[23].Recientemente se han implicado varios genes adicionales en casos raros de PQRAD (p. ej., IFT140, GANAB, NEK8 y DNAJB11 ). El trasfondo poligénico que acompaña a la proteína mutante puede explicar, en parte, la variación interfamiliar observada en la gravedad de la PQRAD[24]. Aunque los mecanismos patogénicos de la PQRAD no están claros, los datos de los modelos de ratón apuntan a una cistogénesis dependiente de la dosis a través de la proteína mutante codificada[25,26].
En los niños, la PQRAD puede diagnosticarse después de que se detecten incidentalmente quistes renales solitarios o múltiples asintomáticos en una ecografía renal. También se han descrito casos de cardiopatía valvular[27] y enfermedad renal quística temprana grave debida a variantes bialélicas de PKD1 o PKD2[28,29]. La enfermedad renal poliquística autosómica recesiva debida a variantes en el gen que codifica la fibrocistina (PKHD1) es principalmente una enfermedad renal quística infantil, pero se han desarrollado casos raros en la edad adulta[30]. Estas presentaciones tardías a menudo dependen de la gravedad de las variantes de PKHD1[31], y con frecuencia se detectan signos extrarrenales de afectación hepática[30].
Hasta hace poco, rara vez se realizaban pruebas genéticas clínicas de rutina en personas con PQRAD[32]. No obstante, un diagnóstico a nivel genético es importante, ya que las variantes en más de 15 genes diferentes pueden imitar un fenotipo de PQRAD . Utilizando la secuenciación del exoma, Chang et al.[33]informaron que 19 de 235 pacientes (8,1%) con un diagnóstico clínico de PQRAD tenían variantes en genes distintos de PKD1 o PKD2. Por lo tanto, las pruebas genéticas en pacientes con enfermedad renal quística pueden informar el manejo clínico y la planificación familiar y reclasificar a los pacientes en el espectro de la PQRAD. Las pruebas genéticas que utilizan Sanger o la secuenciación del exoma de lectura corta de PKD1 pueden tener limitaciones importantes debido a las duplicaciones y la homología con pseudogenes similares[34]. Por lo tanto, la validación a través de un ensayo de reacción en cadena de la polimerasa (PCR) de largo alcance es necesaria para la detección completa de las variaciones de PKD1.
Nefronoptisis
La nefronoptisis es un grupo de enfermedades autosómicas recesivas genéticamente heterogéneas que se caracterizan por un deterioro progresivo e inespecífico de la función renal. El trastorno puede ocurrir a cualquier edad (infancia, adolescencia o adultez) y se caracteriza por poliuria, retraso del crecimiento (en niños) y anemia, pero con un sedimento urinario generalmente insulso. Las manifestaciones extrarrenales, con anomalías oculares, cerebrales, hepáticas o esqueléticas, están presentes en el 15 al 50% de los casos, dependiendo del gen involucrado[35,36].
La nefronoptisis se diagnostica con mayor precisión mediante pruebas genéticas (con un rendimiento de aproximadamente el 70% en casos sospechosos)[37]. Las variantes homocigóticas (más comúnmente deleciones) en NPHP1 representan el 20 al 50% de todos los casos[35,38]. Los casos restantes son causados por variantes en cualquiera de aproximadamente 90 genes diferentes relacionados con múltiples vías moleculares que regulan la polaridad celular plana, la señalización de Sonic Hedgehog, la respuesta al daño del ADN o la señalización de AMP cíclico[36,37]. Los estudios de correlación genotipo-fenotipo muestran que las variantes nulas en los genes de la nefronoptisis se asocian con una edad más joven en la presentación y fenotipos de enfermedad más graves[35,39]. Los datos de una cohorte reciente de 600 pacientes con nefronoptisis sugieren que la enfermedad de inicio en la edad adulta con El subdiagnóstico es la regla[35].
Enfermedades glomerulares genéticas
Las enfermedades glomerulares genéticas suelen implicar productos genéticos mutantes que normalmente mantienen la función de los podocitos o variantes patógenas de las proteínas que componen la membrana basal glomerular (p. ej., colágeno tipo IV). Las presentaciones clínicas incluyen el síndrome nefrótico resistente a esteroides (SNRE) con glomeruloesclerosis focal y segmentaria (GEFS) en la biopsia renal o proteinuria crónica con o sin hematuria. La GEFS es una lesión inespecífica que representa un patrón de lesión de los podocitos en lugar de una entidad patológica definida. Las formas genéticas de la GEFS pueden ser familiares o esporádicas. Pueden estar presentes características extrarrenales, dependiendo del gen involucrado[40].
ERC relacionada con el colágeno tipo IV
Las variantes en los genes que codifican las cadenas α3, α4 y α5 del colágeno tipo IV (COL4A3, COL4A4 y COL4A5, respectivamente) son la segunda causa genética más común de ERC después de la PQRAD, y representan aproximadamente el 2 al 3 % de los casos en adultos con ERC avanzada[8]. Hace más de 30 años, se identificaron alteraciones en el colágeno tipo IV, un componente principal de la membrana basal glomerular, en pacientes con síndrome de Alport (la llamada nefritis familiar). Estudios posteriores han demostrado una amplia variación fenotípica entre pacientes con variantes de colágeno, a menudo asociadas con modos de herencia y la ubicación y tipos de variante dentro de los genes que codifican el colágeno tipo IV. Las características clínicas varían desde el síndrome de Alport clásico (hematuria glomerular, pérdida de audición y anomalías oculares) hasta la enfermedad confinada únicamente a los riñones, con una membrana basal delgada que se observa en la microscopía electrónica, o FSGS. Múltiples estudios muestran que las variantes previamente subestimadas en los genes que codifican el colágeno tipo IV son responsables de muchos casos de ERC en adultos que no tienen las características extrarrenales clásicas del síndrome de Alport[8-11]. Estos pacientes pueden presentar inicialmente hematuria o proteinuria persistentes y aisladas, con el desarrollo posterior de ERC progresiva.
Las correlaciones genotipo-fenotipo para los genes que codifican el colágeno tipo IV se han descrito ampliamente[41-43]. La enfermedad ligada al cromosoma X es causada por variantes patogénicas en COL4A5 y herencia autosómica recesiva o autosómica dominante de variantes patogénicas en COL4A3 o COL4A4. La enfermedad ligada al cromosoma X y autosómica recesiva se asocian con el mayor riesgo de insuficiencia renal. La insuficiencia renal se presenta en la mayoría de los hombres y en el 15 a 30% de las mujeres con enfermedad ligada al cromosoma X, así como en la mayoría de las personas con enfermedad autosómica recesiva[41,42]. El síndrome de Alport resultante de variantes patogénicas en dos genes diferentes que codifican el colágeno tipo IV (herencia digénica) puede tener peores resultados clínicos que la enfermedad debida a una variante heterocigótica de un solo gen[44]. Además, las variantes truncadas se asocian con peores resultados renales, en comparación con las variantes sin sentido, con una edad más temprana al inicio de la insuficiencia renal[41,43]. Las variantes sin sentido a menudo afectan los residuos de glicina que son necesarios para ensamblar la estructura del heterotrímero de colágeno. Establecer el diagnóstico molecular preciso de las variantes de los genes que codifican el colágeno tipo IV es fundamental para la selección del tratamiento adecuado y un posible donante de riñón[45].
Formas monogénicas de síndrome nefrótico autosómico recesivo y FSGS
Hace dos décadas, la identificación de variantes en los genes asociados a los podocitos que codifican la nefrina (NPHS1)[46] y la podocina (NPHS2)’47] en niños con formas congénitas del síndrome nefrótico autosómico recesivo estableció la enfermedad de los podocitos como causa de proteinuria crónica e insuficiencia renal progresiva[48]. Las alteraciones hereditarias autosómicas dominantes y recesivas en más de 50 productos genéticos diferentes que mantienen la ultraestructura de los podocitos, median la transducción de señales o controlan los reordenamientos del citoesqueleto de los podocitos se han descrito como causas genéticas del síndrome nefrótico o la proteinuria crónica[40]. Las personas con estas variantes se presentan con mayor frecuencia durante la infancia, pero ocasionalmente se presentan durante la edad adulta. Por ejemplo, las variantes en la podocina, la proteína mutada más comúnmente en niños con síndrome nefrótico infantil, también se han descrito en casos de proteinuria o FSGS de inicio en la edad adulta. Además, la variante de podocina R229Q, presente en aproximadamente el 4% de la población europea, se ha asociado con un mayor riesgo de FSGS de inicio en la edad adulta, un riesgo modulado por la variante de podocina del segundo alelo[49,50]. Otras causas monogénicas de proteinuria de inicio en la edad adulta y FSGS incluyen variantes autosómicas dominantes en los genes citoesqueléticos ACTN4[51] e INF2[52] y la proteína del canal de cationes codificada por TRPC6[53,54]. La identificación de los síndromes monogénicos asociados puede informar el manejo clínico.
Enfermedades tubulointersticiales
Las afecciones genéticas que afectan la función tubular renal (tubulopatías) abarcan más de 50 síndromes, que generalmente involucran canales iónicos o transportadores[55]. Las características clínicas pueden incluir desequilibrios en el desplazamiento de líquidos. Las anomalías de laboratorio incluyen alteraciones electrolíticas o ácido-base y proteinuria tubular. Las imágenes pueden mostrar nefrolitiasis o nefrocalcinosis, y a menudo se observa daño tubulointersticial inespecífico en muestras de biopsia renal. La uromodulina (también conocida como proteína de Tamm-Horsfall) es una proteína específica del riñón que se sintetiza en la rama ascendente gruesa del asa de Henle y se secreta en la orina. Es la proteína más abundante en la orina normal y está involucrada en la regulación del transporte de sal, la defensa contra infecciones del tracto urinario y la formación de cálculos[56]. Los estudios sugieren que la enfermedad autosómica dominante debido a variantes de ganancia de función en UMOD es una de las causas monogénicas más prevalentes de ERC en adultos en todo el mundo, y representa aproximadamente el 0,3 al 1 % de todos los casos de ERC avanzada[8,57,58].
Las variantes de UMOD59 causan un espectro de trastornos que se han denominado enfermedad renal tubulointersticial autosómica dominante relacionada con UMOD (ADTKD)[56]. La ADTKD se caracteriza por una insuficiencia renal insidiosa entre la tercera y la sexta décadas de la vida. Los pacientes también pueden tener hiperuricemia y gota relacionadas con la excreción fraccional reducida de urato, a pesar de una TFG normal[58]. Además de la enfermedad tubulointersticial que es evidente en la biopsia renal, se han informado características de FSGS[60]. A menudo, la ADTKD pasa desapercibida porque carece de características clínicas o histológicas distintivas; de hecho, pocos médicos conocen la entidad de la enfermedad y las pruebas genéticas a menudo no están disponibles. La mayoría de las variantes genéticas causantes de enfermedades son variantes sin sentido, a menudo ubicadas en los exones 3 y 4; el 60% involucra un residuo de cisteína[58]. Las variantes patógenas de UMOD causan un plegamiento incorrecto de proteínas, con la posterior retención de proteínas en el retículo endoplasmático y la orientación incorrecta de la uromodulina en la rama ascendente gruesa del asa de Henle. El daño tubulointersticial resultante conduce a la ERC[61].
Las variantes en otros genes también pueden causar ADTKD. Las variantes en MUC1 son la segunda causa genética más común de ERDADT y siempre deben considerarse en casos negativos a UMOD. Se requieren pruebas especializadas para identificar las variantes de MUC1[62], que son indetectables con la secuenciación de exomas de Sanger o de lectura corta. Además, las variantes heterocigóticas raras en REN[63,64] o SEC61A1[65] pueden dar lugar al fenotipo ERDADT.
Enfermedad de cálculos renales
La enfermedad de cálculos renales es una afección multifactorial que a menudo incluye un componente genético. En muchos casos, la enfermedad es impulsada por desequilibrios metabólicos que conducen a la cristalización urinaria. Por lo tanto, se espera que las variantes patógenas en genes que codifican productos que normalmente mantienen el equilibrio metabólico causen formas hereditarias de nefrolitiasis, que a veces conducen a la enfermedad renal crónica. Se han implicado más de 30 genes diferentes en la formación de cálculos y se ha estimado que afectan del 16 al 29% de los niños y el 11% de los adultos con nefrolitiasis[66-68]. Aunque el porcentaje general de pacientes con insuficiencia renal que tienen nefrolitiasis es solo del 3%, las formas genéticas de la enfermedad de cálculos renales, como la deficiencia de adenina fosforribosiltransferasa, la enfermedad de Dent, la hipomagnesemia familiar con hipercalciuria y nefrocalcinosis y la hiperoxaluria primaria, con frecuencia conducen a la enfermedad renal crónica y progresan a la insuficiencia renal. Además, los pacientes con deficiencia de adenina fosforribosiltransferasa o hiperoxaluria primaria corren el riesgo de sufrir una recurrencia de la enfermedad después del trasplante renal, ya que el trasplante no corrige el defecto metabólico subyacente.
Enfermedad renal crónica de causa desconocida
En aproximadamente el 10 al 20 % de los casos de insuficiencia renal en adultos en los Estados Unidos y Europa, la causa de la enfermedad renal crónica sigue siendo desconocida después de un estudio exhaustivo, que incluye imágenes y biopsias renales[69]. Los informes sugieren que hasta el 20 % de estos casos pueden atribuirse a afecciones genéticas[8,9,70]. Aunque a menudo se detectan variantes en los genes que codifican el colágeno tipo IV (en hasta el 30 % de los casos), un panel completo de genes renales o la secuenciación del exoma pueden ser la primera prueba diagnóstica preferida, dado que muchos casos no están relacionados con variantes de genes de colágeno.
Otros aspectos de la enfermedad renal crónica monogénica
Los síndromes monogénicos raros que pueden causar daño renal secundario también deben considerarse en una investigación de las causas genéticas subyacentes de la enfermedad renal crónica. Estos síndromes incluyen diabetes monogénica, hiperlipidemia o hipertensión monogénica y lupus eritematoso sistémico monogénico[71].
Nefropatías con una base genética compleja
ERC relacionada con APOL1
APOL1 pertenece a una familia de proteínas que confieren protección contra infecciones con enfermedad del sueño africana mediada por tripanosoma. En 2010, dos grupos de investigadores identificaron de forma independiente dos conjuntos de variantes de APOL1 (un par vinculado de variantes sin sentido, S342G e I384M, y dos deleciones consecutivas de aminoácidos, N388 e Y389) dentro de una región genómica previamente sospechada[72-74] como impulsores genéticos de varios subtipos de ERC progresiva[75,76]. Estos conjuntos de variantes mutuamente excluyentes, designados G1 y G2, respectivamente, confieren protección contra un espectro extendido de especies de tripanosoma en comparación con el alelo ancestral G0. La hipótesis de la ventaja heterocigótica afirma que ambas variantes aumentaron hasta alcanzar una frecuencia alélica alta en las poblaciones subsaharianas para brindar protección contra la infección por tripanosoma, pero estas variantes contribuyen a la frecuencia aumentada, observada desde hace tiempo, de enfermedad renal crónica entre personas con ascendencia reciente del África subsahariana[77].
APOL1 está asociada con un amplio espectro de nefropatías, incluidas la glomerulonefritis esclerosante focal y segmentaria (GEFS), la nefropatía hipertensiva y la nefropatía asociada al virus de la inmunodeficiencia humana (VIH), así como la nefritis asociada al lupus[78]; estas asociaciones definen un nuevo espectro de enfermedad renal crónica relacionada con APOL1. Por ejemplo, dos variantes de APOL1 confieren un aumento del riesgo de FSGS por un factor de 10 a 17 y un aumento del riesgo de nefropatía asociada al VIH por un factor de 29 a 89[79,80]. Aunque algunos informes han indicado la posibilidad de un riesgo leve de enfermedad renal incluso con un solo alelo de riesgo G1 o G2[81] esto no se ha establecido.
Los estudios in vitro e in vivo[82] respaldan un mecanismo de ganancia de función tóxica en pacientes con dos variantes de APOL1. Aunque los mecanismos exactos de la lesión celular no están completamente dilucidados, el éxito de nuevas terapias como la inaxaplina en la FSGS apunta a los podocitos glomerulares como el objetivo de la lesión celular. Se cree que el tráfico del producto del gen aberrante a través del retículo endoplasmático luminal da como resultado una actividad anormal del canal de cationes de la membrana celular. Este paso próximo de la lesión celular es seguido por una cascada descendente de inflamación y más lesión celular[83,84].
La correlación genotipo-fenotipo en APOL1 parece ser única. Aunque es evidente un modo de herencia causal recesivo (bialélico) (la enfermedad está fuertemente asociada causalmente con los genotipos G1/G1, G2/G2 y G1/G2, no con G0/G0, G1/G0 o G2/G0), la alta penetrancia mendeliana clásica que a menudo se observa en las enfermedades autosómicas recesivas está ausente en la ERC asociada a APOL1. Las variantes G1 y G2 son relativamente comunes, aproximadamente el 13% de los afroamericanos y más de 70 millones de personas en todo el mundo tienen genotipos APOL1 de alto riesgo[85,86]. Sin embargo, la ERC se desarrolla solo en un subgrupo de estas personas (penetración incompleta), dependiendo de algunos factores contribuyentes conocidos (p. ej., infección por VIH o estados altos de interferón[87]) y otros factores desconocidos. Otro refinamiento importante ha sido la reciente observación de que otra variante de la secuencia de ADN, que codifica un cambio de aminoácido de asparagina a lisina (N264K), hace que la variante de riesgo G2 sea inocua en el pequeño porcentaje de personas con el genotipo G2/G2 o G1/G2, que de otro modo sería de alto riesgo[88,89]. Por lo tanto, las pruebas genéticas con fines de investigación y manejo clínico deberían incluir la alteración N264K. Una sólida justificación para las pruebas de APOL1 en los paneles de genes de enfermedades glomerulares y, en particular, para los pacientes de ascendencia africana que tienen enfermedad renal crónica proteinúrica es la capacidad de inscribir a los pacientes en ensayos clínicos en curso de terapias dirigidas a APOL1 (como se señala en la sección siguiente sobre terapias emergentes).
Nefropatía membranosa idiopática y nefropatía por IgA
Los estudios de asociación genómica de gran importancia que se han llevado a cabo durante la última década han investigado la base genética de los casos de nefropatía membranosa idiopática[90,91] y nefropatía por IgA[92,93], comprobados mediante biopsia. Los resultados muestran que cada una de estas glomerulopatías inmunomediadas puede tener un panorama genético revelador con loci de riesgo específicos de la enfermedad que pueden arrojar luz sobre los mecanismos fisiopatológicos . Se requieren más estudios para determinar si estos hallazgos serían útiles en el ámbito clínico.
Evaluación clínica y diagnóstica
El primer paso para establecer un diagnóstico genético es la fenotipificación integral. Obtener una historia familiar detallada y un pedigrí de tres generaciones también son pasos importantes. No obstante, una historia familiar negativa de enfermedad renal no excluye la ERC de origen genético. El rendimiento diagnóstico de las pruebas genéticas en la ERC es variable y depende del fenotipo de la enfermedad, las condiciones coexistentes asociadas, la etnia, la consanguinidad, los antecedentes familiares y la edad de inicio. Múltiples estudios respaldan una lista de indicaciones importantes que deberían impulsar a los médicos a considerar el análisis genético para sus pacientes con ERC . Además, varios informes han indicado que el uso de criterios establecidos puede dar como resultado un alto rendimiento diagnóstico general de las pruebas genéticas en la ERC (40 a 57 %), generalmente en clínicas de nefrología centradas en diagnósticos genéticos y en clínicas genéticas[94-96]. Descubrir la causa genética subyacente de la ERC puede mejorar la precisión del diagnóstico y contribuir a un seguimiento y tratamiento específicos. También puede obviar la necesidad de procedimientos de diagnóstico más invasivos y permitir un asesoramiento genético preciso para la planificación familiar. Si se indica un análisis genético, los pacientes y sus familias deben recibir asesoramiento genético antes y después de la prueba para recibir información precisa sobre la enfermedad sospechada, la técnica de prueba genética recomendada, el rendimiento diagnóstico estimado y las posibles implicaciones, incluida la posibilidad de hallazgos genéticos incidentales. La prueba genética específica más apropiada depende de la presentación clínica particular del paciente, la disponibilidad local de diagnósticos moleculares y la preferencia del paciente. Los análisis de genes de enfermedad renal individual están siendo reemplazados actualmente por grandes paneles de genes, secuenciación del exoma y secuenciación del genoma completo.
Una vez que se reciben los resultados de las pruebas genéticas, incluso si son negativos, es importante realizar una consulta genética para realizar una reevaluación clínica y considerar la posibilidad de realizar pruebas genéticas más amplias o complementarias. A menudo se requieren pasos de diagnóstico adicionales y es importante interpretar correctamente los resultados. La interpretación de las variantes genéticas debe basarse en las pautas internacionalmente aceptadas de la American College of Medical Genetics and Genomics–Association for Molecular Pathology[98]. Los pacientes con resultados negativos de las pruebas genéticas deben someterse a una reevaluación periódica.
La interpretación de las variantes genéticas debe realizarse con el uso de controles con ascendencia coincidente, si están disponibles[99]. Sin embargo, muchas poblaciones en las que se presenta la ERC están subrepresentadas en las bases de datos genéticas y los exomas específicos de la población se han interpretado con frecuencia en el contexto de datos de referencia europeos o asiáticos. Esto puede conducir a un sesgo de ascendencia, que puede dar lugar a una atribución incorrecta de la patogenicidad de una variante y la consiguiente interpretación clínica errónea[9].
Terapias emergentes
La prevención primaria, secundaria y terciaria de la ERC es cada vez más posible gracias a los diagnósticos genéticos. Un diagnóstico genético establece la elegibilidad para terapias eficaces relacionadas con los genes y permite que los pacientes se conviertan en candidatos para los tratamientos emergentes.
Un ejemplo reciente involucró un estudio de fase 2a a corto plazo del uso de una molécula pequeña (inaxaplin) para tratar la ERC relacionada con APOL1.100 La molécula revierte el efecto tóxico de ganancia de función de las variantes de alto riesgo de APOL1. Este estudio clínico temprano involucró a participantes con dos variantes de APOL1 y FSGS comprobada por biopsia. La mayoría de los participantes tuvieron una reducción clínicamente significativa de la proteinuria después de 13 semanas de tratamiento.100 El estudio proporcionó evidencia inicial de que la inhibición dirigida de la función de APOL1 es efectiva y puede detener la progresión de la ERC relacionada con APOL1. Se está llevando a cabo un ensayo de fase 2-3 (número NCT05312879 en ClinicalTrials.gov).
La identificación de enfermedades monogénicas de la enfermedad renal crónica puede facilitar estrategias terapéuticas moleculares dirigidas. Por ejemplo, un estudio reciente en un modelo de ratón mostró que la reexpresión de la proteína policistina 1 a través de la transferencia del transgén Pkd1 retrasó notablemente el desarrollo de quistes, retrasó la progresión de los quistes y pospuso la insuficiencia renal en ratones sin Pkd1.101 Otro enfoque prometedor utiliza tecnologías basadas en ARN, incluidos oligonucleótidos antisentido modificados químicamente o pequeños ARN interferentes, que modulan la abundancia, el procesamiento y la producción de traducción del ARN celular sin la necesidad de la administración del transgén al riñón, que es más complejo.
Estos agentes basados en ARN pueden cambiar la expresión de cualquier proteína, incluso proteínas que no son susceptibles a los enfoques tradicionales que involucran moléculas pequeñas, y pueden usarse para objetivos que anteriormente no eran “medicables”. Por ejemplo, un modelo murino del síndrome de Alport sugiere que la terapia de omisión de exón con el uso de oligonucleótidos antisentido dirigidos a variantes truncadas en el exón 21 de COL4A5 es beneficiosa para retardar la progresión del daño renal.102 Esta estrategia ha dado como resultado una expresión fenotípica más leve al evitar un codón de terminación temprano. De manera similar, el tratamiento con oligonucleótidos antisentido mejoró la proteinuria inducida por interferón-γ en la nefropatía relacionada con APOL1 con el uso de un modelo murino transgénico.103 Una aplicación reciente y exitosa de esta tecnología en humanos implicó el uso de un tratamiento con ARN interferente pequeño (lumasiran y nedosiran) para la sobreproducción hepática de oxalato en pacientes con hiperoxaluria tipo 1 debido a variantes AGXT.104,105 Lumasiran redujo la excreción urinaria de oxalato, que generalmente causa insuficiencia renal en estos pacientes.
Sobre la base de estos conceptos, se están estudiando tratamientos genéticos adicionales para enfermedades renales. La administración de fármacos a las células renales específicas de interés sigue siendo un desafío.
Conclusiones y direcciones futuras
El uso de pruebas genéticas en el manejo de la enfermedad renal crónica y en los ensayos de tratamiento clínico está aumentando rápidamente y, en última instancia, puede transformar la práctica nefrológica mediante el desarrollo de terapias específicas para genes. Se necesita más trabajo para definir el papel de la genética en los donantes de riñón y para desarrollar tratamientos adicionales específicos para genes. Muchas enfermedades renales genéticas son raras. Las cohortes de estudios internacionales aumentarán la información genética de poblaciones subrepresentadas y diversas con enfermedades renales y deberían ayudar a aliviar la gran carga mundial de la enfermedad renal crónica.
Figure 1. Riesgo de enfermedad renal crónica genética (ERC) según la penetrancia genética y los factores contribuyentes.
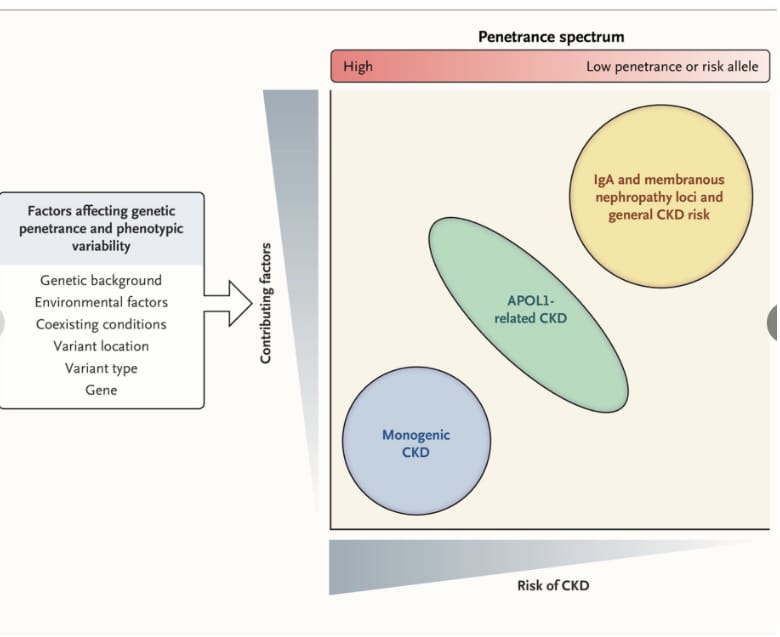
La penetrancia genética se refiere a la proporción de personas con una variante genética particular que tienen un fenotipo de enfermedad atribuido a la variante genética. La terminología genética de consenso ha sido definida recientemente por el National Institutes of Health Clinical Genome Resource (ClinGen).19 La “penetrancia baja” describe variantes para las cuales puede haber evidencia de segregación en un patrón de herencia mendeliano pero sin el desarrollo de características de la enfermedad en la mayoría de los portadores. Sin embargo, los portadores de estas variantes tienen una tasa más alta de enfermedad que la prevalencia de fondo para la condición asociada, lo que hace que la presencia de una variante de baja penetrancia por sí sola sea clínicamente significativa, como es evidente en el caso de la ERC relacionada con APOL1. El “alelo de riesgo” describe variantes que no son en sí mismas causales y no se manifiestan en un patrón de herencia mendeliano pero que pueden combinarse con otras variantes genómicas para aumentar el riesgo de ERC. Además, un alelo de este tipo, en lugar de causar una enfermedad, puede estar genéticamente vinculado a otra variante que contribuya directamente a la causa de la enfermedad o puede ser parte de un conjunto de variantes para las cuales no se puede distinguir una única variante principal que contribuya al riesgo de enfermedad. Algunos ejemplos son los alelos de riesgo vinculados a la nefropatía por IgA o la nefropatía membranosa idiopática.
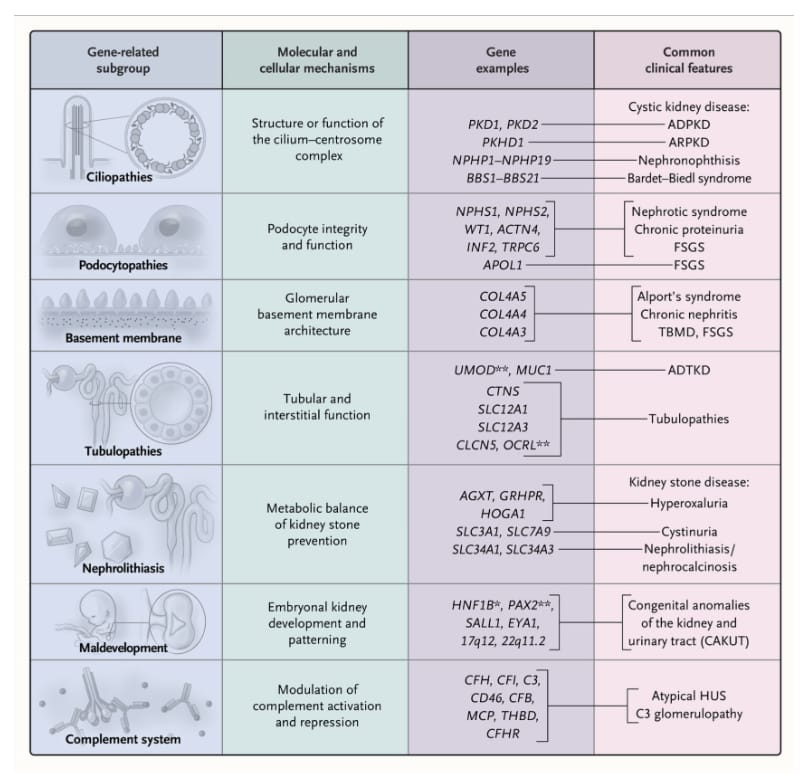
Principales categorías celulares y moleculares de la ERC de origen genético.
Se muestran los genes más comunes y sus diversas características clínicas como ejemplos de síndromes relacionados con la ERC de origen genético en cada subgrupo mostrado. Dada la magnitud de la heterogeneidad genética y fenotípica, combinada con la importancia de la delimitación precisa de las variantes causales para el manejo de la enfermedad, el uso de la categorización de la ERC basada en los genes se está convirtiendo en un estándar de la práctica clínica. Los trastornos relacionados con la ERC de origen genético se pueden definir mediante variantes genéticas específicas que brindan información molecular y mecanicista sobre las características fisiopatológicas y sobre las posibles opciones de tratamiento. Por lo tanto, las condiciones de ERC similares se pueden manejar con tratamientos que difieren según la patogenia molecular específica. Las formas genéticas de la ERC se caracterizan por altos grados de heterogeneidad genética y fenotípica. Por ejemplo, un diagnóstico histológico de glomeruloesclerosis focal y segmentaria (GEFS) no es específico y refleja un proceso fisiopatológico compartido y un resultado histopatológico posterior a muchas variantes de un solo gen diferentes, como las de APOL1, COL4A5, UMOD e INF2. Cada variante inicia un proceso causal distinto, que finalmente conduce a la ERC con hallazgos de GFS en la biopsia renal. Este fenómeno se conoce como heterogeneidad genética o de locus. Por el contrario, las variantes en el mismo gen a veces pueden conducir a diferentes fenotipos clínicos e histopatológicos de ERC entre pacientes individuales. Por ejemplo, las variantes en HNF1B pueden conducir a anomalías renales del desarrollo, así como disfunción tubular que conduce a hipomagnesemia e hiperuricemia, o causar enfermedad renal quística, que puede ser fenotípicamente similar a la enfermedad renal poliquística autosómica dominante (ERPAD). Este fenómeno se conoce como heterogeneidad alélica o fenotípica. Las líneas horizontales representan fenotipos clásicos. Se ha informado en raras ocasiones que el gen HNF1B (un asterisco) causa un fenotipo de enfermedad renal tubulointersticial autosómica dominante (ADTKD). También se ha informado que los genes CLCN5, OCRL y PAX2 (doble asterisco) causan FSGS. ARPKD denota enfermedad renal poliquística autosómica recesiva, síndrome hemolítico-urémico HUS.