Aspectos destacados
- Tres subtipos moleculares de cáncer de próstata revelados mediante un análisis proteómico integral
- Un panel de 16 proteínas predice eficazmente la BCR para pacientes con cáncer de próstata en diferentes etapas
- La inhibición de NUDT5 y SEPTIN8 suprime las características malignas del cáncer de próstata
Resumen
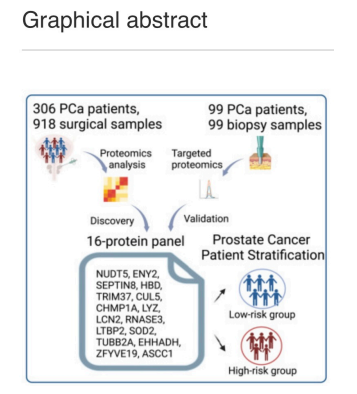
El cáncer de próstata (CaP) es el tumor maligno más común en los hombres. Actualmente, existen pocos indicadores de pronóstico para predecir los resultados del CaP y guiar los tratamientos. Rui Sun,et.al.,[Cell Reports Medicine. Volume 5, Issue 8101679August 20, 2024] realizaron un perfil proteómico integral de 918 muestras de tejido de 306 pacientes chinos con CaP utilizando espectrometría de masas de adquisición independiente de datos (DIA-MS). Identificaron más de 10 000 proteínas y definieron tres subtipos moleculares de CaP con diferencias clínicas y proteómicas significativas.
=> Recibir por Whatsapp las noticias destacadas
Desarrollaron un panel de 16 proteínas que predice eficazmente la recurrencia bioquímica (BCR) para pacientes con cáncer de próstata, que se valida en seis conjuntos de datos publicados y una cohorte adicional de 99 muestras de biopsia mediante proteómica dirigida.
Curiosamente, este panel de 16 proteínas predice eficazmente la BCR en diferentes grados y estadios patológicos de la Sociedad Internacional de Patología Urológica (ISUP) y supera al sistema de clasificación de riesgo de D’Amico en la predicción de BCR. Además, la doble eliminación de NUDT5 y SEPTIN8, dos componentes del panel de 16 proteínas, suprime significativamente la proliferación, invasión y migración de las células de cáncer de próstata, lo que sugiere que la combinación de NUDT5 y SEPTIN8 puede proporcionar nuevos enfoques para el tratamiento del cáncer de próstata.
En Detalle
El cáncer de próstata (PCa) es el tipo de cáncer más común y la segunda causa principal de muertes relacionadas con el cáncer entre los hombres en todo el mundo. En las regiones médicamente avanzadas, la proporción de PCa limitado o localmente progresivo en el momento del diagnóstico inicial está aumentando gradualmente y tiende a dominar la población general diagnosticada. El principal tratamiento curativo para esta población es la prostatectomía radical con terapia adyuvante/neoadyuvante.
La incidencia de recurrencia bioquímica (BCR) después de la prostatectomía radical puede ser tan alta como el 40%, y está fuertemente vinculada con la recurrencia clínica posterior, la metástasis y la muerte específica por cáncer. Como resultado, la predicción del riesgo de BCR utilizando una variedad de indicadores clínicos para dirigir la evaluación clínica es ahora un enfoque ampliamente utilizado en el entorno del tratamiento del PCa.
Estos indicadores suelen incluir la agrupación de la Sociedad Internacional de Patología Urológica (ISUP), el antígeno prostático específico (PSA) en el momento del diagnóstico, el estadio clínico, etc. El método de categorización de riesgo D’Amico más clásico, que integra el grupo ISUP y el valor de PSA en el momento del diagnóstico, ha sido respaldado sistemáticamente por un gran número de expertos de todos los grupos étnicos.
Según el método de clasificación de riesgo D’Amico, los pacientes con CaP se pueden agrupar en grupos de riesgo bajo, intermedio y alto, con riesgos estimados de BCR a 5 años de <25%, 25%–50% y >50%, respectivamente. También se evaluó la eficacia de combinar parámetros radiológicos y clínicos para medir el riesgo de BCR, y se demostró que aumenta la precisión predictiva del método de estratificación del riesgo.
Sin embargo, debido a la amplia detección del PSA del CaP, la distribución de los pacientes cambió gradualmente con el tiempo de los grupos de alto riesgo a los grupos de riesgo bajo e intermedio; La relevancia clínica de este esquema de clasificación puede ser limitada y estar disminuyendo en la actualidad. Además, los métodos de estratificación actuales no logran explicar los mecanismos de progresión del cáncer de próstata y no proporcionan nuevos objetivos terapéuticos para el tratamiento del cáncer de próstata.
Las extensas investigaciones genómicas, epigenómicas y transcriptómicas han propuesto nuevas clasificaciones para el cáncer de próstata basadas en anomalías genómicas específicas, como mutaciones genéticas específicas (SPOP, FOXA1, IDH1, etc.) y fusiones (ERG, ETV1/4, FLI1, etc.).
Sin embargo, las proteínas son los principales ejecutores de los procesos celulares, y las alteraciones en los perfiles genéticos y transcripcionales no siempre son consistentes con las alteraciones en los perfiles o actividades proteicas. Por lo tanto, una estrategia de clasificación y predicción de riesgos basada en proteínas podría proporcionar mejores conocimientos sobre los procesos patológicos, la instrucción terapéutica y la predicción del pronóstico para la investigación del cáncer.
Si bien hasta la fecha se han perfilado miles de genomas y transcriptomas de PCa, los perfiles proteómicos publicados de PCa se limitaron a un pequeño número de muestras de PCa. Un estudio proteogenómico analizó 39 muestras de PCa e identificó una subred de nueve genes que se correlacionaba positivamente con fenotipos tumorales más agresivos y predecía la supervivencia sin recurrencia.
Sinha et al. demostró que los biomarcadores pronósticos basados en proteómica identificados en una cohorte de 76 pacientes con CaP eran superiores a los basados en genómica y transcriptómica. La participación crucial del ciclo del ácido tricarboxílico (TCA) en la progresión del CaP se evidenció mediante proteómica en lugar de transcriptómica a través de un estudio multiómico de 28 pacientes con CaP.
Un estudio proteómico de 28 pacientes con CaP encontró que pro-NPY podría ser un biomarcador pronóstico potencial. Un perfil fosfoproteómico, junto con un análisis genómico y transcriptómico, reveló información de la vía clínicamente relevante potencialmente adecuada para la estratificación de pacientes y terapias dirigidas en el CaP en etapa tardía utilizando 27 muestras. Un estudio proteómico a gran escala de 278 pacientes occidentales con CaP derivó una estrategia de clasificación de riesgo para el riesgo intermedio de CaP.
Sin embargo, el CaP es altamente heterogéneo y existen diferencias conocidas en las alteraciones genéticas del CaP entre diferentes grupos étnicos. La mayoría de los estudios basados en proteómica se realizaron en cohortes relativamente pequeñas de diferentes países occidentales. Nuestro conocimiento de los mecanismos moleculares detrás de la progresión del cáncer de próstata y la BCR aún es insuficiente, especialmente en la población china.
Por lo tanto, es urgente realizar un análisis proteómico integral de una gran cohorte de pacientes con cáncer de próstata: podría proporcionar una clasificación mucho más precisa de los pacientes con cáncer de próstata y nuevos conocimientos sobre sus mecanismos moleculares.
El análisis proteómico basado en espectrometría de masas de adquisición independiente de datos (DIA-MS) proporciona una amplia gama de aplicaciones para explorar nuevos biomarcadores y objetivos terapéuticos en varios campos médicos debido a su alto rendimiento y ventajas de cuantificación precisa.
En particular, la fragmentación serial de acumulación paralela combinada con tecnología de adquisición independiente de datos es una tecnología proteómica de vanguardia que logra una mayor profundidad de identificación del proteoma mediante la reducción de la interferencia de ruido y el aumento de la sensibilidad de la señal.
Rui Sun,et.al.,[Cell Reports Medicine. Volume 5, Issue 8101679August 20, 2024] utilizaron la tecnología DIA-MS para caracterizar el panorama del proteoma del PCa utilizando 918 muestras de 306 pacientes chinos con PCa que se sometieron a cirugía de prostatectomía radical (PCSHA). Midieron más de 10,000 proteínas, mapearon las características proteómicas asociadas con diferentes grupos ISUP e identificaron tres subtipos de PCa con diferencias clínicas y moleculares significativas.
En particular, identificaron un panel de 16 proteínas que predicen eficientemente BCR, lo que supera las estrategias que se utilizan actualmente en la práctica clínica. Este modelo se validó de manera efectiva en seis conjuntos de datos externos.
Además, se validó en una cohorte china independiente que comprendía 99 muestras de biopsia cuantificadas mediante una tecnología proteómica dirigida. Entre estas 16 proteínas, destacamos NUDT5 y SEPTIN8 como candidatos prometedores para dianas sintéticas en el tratamiento del cáncer de próstata. Este conjunto de datos proteómicos integral llena el vacío en el campo de los estudios proteómicos del cáncer de próstata chino al revelar nuevos mecanismos moleculares, métodos de predicción pronóstica y posibles estrategias terapéuticas para el cáncer de próstata.
El recurso proteómico integral de los pacientes chinos con cáncer de próstata
En este estudio, utilizando la tecnología DIA-MS, generaron un panorama proteómico en profundidad, muy valioso y completo del cáncer de próstata. En total, se identificaron 10 045 proteínas en 918 tejidos de próstata FFPE de 306 pacientes chinos con cáncer de próstata. Con el uso de la tecnología de IA, los datos proteómicos altamente reproducibles producidos por las técnicas proteómicas basadas en DIA pueden investigarse más a fondo para identificar más proteínas y obtener información importante adicional.
Los pacientes con cáncer de próstata incluidos en investigaciones proteómicas basadas en espectrometría de masas anteriores son de países occidentales. Por lo tanto, hasta donde sabemos, este conjunto de datos proteómicos, que incluye la mayor cantidad de pacientes con cáncer de próstata y la mayor cantidad de proteínas que se han identificado hasta ahora, es el más grande y completo, por lo que sirve como un recurso proteómico valioso para el campo del cáncer de próstata.
Para tener en cuenta la heterogeneidad intratumoral, recolectaron al menos dos muestras tumorales de cada paciente. Sin embargo, observaron que la heterogeneidad intratumoral era mucho menor que la intertumoral, lo que destaca la importancia de incluir a una gran cantidad de pacientes para comprender completamente los mecanismos subyacentes del CaP.
A diferencia de otras investigaciones a gran escala sobre las proteínas del CaP, esta cohorte se distingue por incluir una gran cantidad de pacientes chinos con CaP. A excepción de las vías de desregulación comunes, como el aumento de la capacidad de fosforilación oxidativa, la desregulación del ciclo del TCA y la disfunción mitocondrial en muestras tumorales, se confirma la convergencia de mutaciones diferentes en la vía común. La investigación también descubrió varias vías distintas enriquecidas en tumores, incluido el metabolismo del RNA, la síntesis de ribosomas, la traducción mitocondrial y la digestión metabólica de aminoácidos.
La taxonomía molecular de los pacientes con CaP
Dada la amplia heterogeneidad del CaP, la subtipificación molecular podría ayudar a explorar los mecanismos divergentes de la enfermedad y predecir resultados clínicos y tratamientos variables. La genómica se ha utilizado para identificar subtipos moleculares de pacientes con CaP basándose en mutaciones genéticas específicas. Sobre la base de este genotipo clásico basado en mutaciones, se han propuesto más clasificaciones incorporando datos transcriptómicos, parámetros clínicos y otros factores.
Rui Sun,et.al.,[Cell Reports Medicine. Volume 5, Issue 8101679August 20, 2024] propone una nueva subtipificación de pacientes con CaP (CPC1, CPC2 y CPC3) basada en el proteoma de su cohorte y demostraron que concuerda con el pronóstico de BCR observado de los pacientes.
CPC3 tuvo el peor pronóstico y se caracterizó por la regulación negativa de los procesos relacionados con el metabolismo y los procesos celulares involucrados en la interacción célula-célula y la regulación positiva del procesamiento de la información genética y las vías de señalización del receptor de células inmunes.
Algunas de estas vías de señalización se observaron en estudios previos. En las vías del metabolismo de aminoácidos, se ha informado que el metabolismo de la arginina, la glicina, la serina y los ácidos grasos desempeñan papeles importantes en la progresión del PCa.
Se ha encontrado la mutación FOXA1 en pacientes chinos con PCa,12 que es un factor pionero para regular la unión del DNA por distintos factores de transcripción y empalme de RNA. Por lo tanto, el procesamiento de la información genética podría ser más venerable en esta cohorte PCSHA, lo que sugiere nuevos conocimientos para la guía del tratamiento del PCa y la predicción del pronóstico.
En este trabajo, los grupos moleculares están vinculados al pronóstico del PCa , lo que podría informar a los médicos sobre las características moleculares del PCa con riesgo bajo o alto de BCR, y proporcionar orientación a los médicos para desarrollar un plan de tratamiento personalizado para el PCa con diferentes características moleculares.
Un panel de 16 proteínas para la predicción de BCR
Los autores desarrollaron un panel de 16 proteínas capaz de predecir BCR de pacientes con cáncer de próstata. El modelo demostró un rendimiento de predicción de BCR superior al de otros sistemas de predicción comúnmente empleados basados en el nivel de PSA, grado ISUP, estadio patológico o clasificación de riesgo de D’Amico .
Es importante destacar que el modelo de 16 proteínas puede predecir eficazmente BCR en diferentes grados ISUP y estadios patológicos, lo que sugiere su posible aplicación para predecir BCR incluso en las primeras etapas del cáncer de próstata.
Se utilizaron el conjunto de datos de validación de PCSHA, así como otros seis conjuntos de datos de cáncer de próstata publicados, incluidos tres conjuntos de datos transcriptómicos y tres conjuntos de datos proteómicos, para evaluar la eficacia y la generalización del modelo de 16 proteínas en la predicción de BCR.
De manera similar a su poder de predicción de BCR en el conjunto de datos de descubrimiento de PCSHA, el modelo de 16 proteínas predijo eficazmente BCR para pacientes con PCa tanto en el conjunto de datos de validación de PCSHA como en los tres conjuntos de datos transcriptómicos .
Sin embargo, la capacidad de predicción de pronóstico del panel de 16 proteínas fue menos efectiva en los tres conjuntos de datos proteómicos (con AUC menores a 0,7), lo que podría atribuirse potencialmente a la menor profundidad de detección y la mayor tasa de omisión observada en los conjuntos de datos proteómicos publicados, así como a los tamaños de cohorte más pequeños en comparación con la cohorte actual. Es importante enfatizar que una evaluación y validación robustas requieren una cantidad adecuada de muestras.
Zhong et al. desarrollaron un panel de 18 proteínas (HR = 2,72) para la predicción de pronóstico utilizando datos proteómicos. El HR es menor que el de este estudio (HR = 3,79). Sin embargo, cuando su modelo de 18 proteínas se aplicó al conjunto de datos PCSHA para la predicción de BCR, se encontró que su AUC era 0,768, menor que la deL panel de 16 proteínas.
Dada la variabilidad significativa en la epidemiología y los antecedentes genéticos del PCa entre diferentes grupos étnicos, la eficacia del panel de predicción de BCR de 18 proteínas para pacientes chinos con PCa puede ser limitada. En consecuencia, estas dos cohortes de diferentes etnias se complementan entre sí, expandiendo el panorama proteico del PCa y brindando recursos valiosos para que los investigadores comprendan mejor el PCa.
Además, se utilizó proteómica dirigida a PRM para confirmar la eficacia de este panel de 16 proteínas en la predicción de BCR en una cohorte de biopsia independiente de 99 pacientes con PCa. La eficacia de predicción de BCR del modelo de 16 proteínas en muestras de biopsia fue similar a la de las muestras de cirugía radical , lo que demuestra la solidez y la eficacia de este panel de 16 proteínas en la predicción de BCR para pacientes con PCa en varias etapas.
Además, en base a estos hallazgos, puede ser posible emplear este panel de 16 proteínas para evaluar el pronóstico del cáncer de próstata antes de la prostatectomía radical. Esto no solo brindaría a los pacientes con cáncer de próstata más información para ayudarlos a tomar decisiones sobre su plan de tratamiento posterior, sino que también brindaría a los médicos una mejor orientación sobre cómo tratar a los pacientes con cáncer de próstata de alto riesgo de manera rápida y agresiva.
Cabe destacar que, tanto en las muestras de cirugía radical como de biopsia, la eficacia predictiva de este panel de 16 proteínas para BCR supera con creces la del método de predicción de D’Amico más utilizado, lo que sugiere que el panel de proteína 16 podría reemplazar potencialmente a D’Amico para la predicción de BCR en la clínica. Sin embargo, este modelo aún necesita ser validado en cohortes clínicas más grandes antes de que pueda traducirse más en la clínica.
Aunque la espectrometría de masas de alta resolución actualmente no es directamente aplicable a las pruebas clínicas de rutina, la espectrometría de masas dirigida podría transferirse a las pruebas clínicas en un futuro cercano. Mientras tanto, la medición de la expresión de estas 16 proteínas mediante inmunohistoquímica podría implementarse en la clínica. Por lo tanto, el panel de predicción de 16 proteínas, que es fácil de implementar, altamente operativo y accesible, se puede utilizar para predecir el pronóstico del cáncer de próstata en el ámbito clínico. Se puede utilizar para mejorar la eficacia diagnóstica, así como en el proceso de toma de decisiones para encontrar objetivos y opciones terapéuticas.
Posibles dianas terapéuticas para el cáncer de próstata
El hallazgo de que muchas proteínas que pueden tratarse con fármacos están reguladas a la baja en pacientes con mal pronóstico no solo explica en cierta medida la disponibilidad limitada de fármacos específicos para el cáncer de próstata, sino que también sugiere nuevas ideas para nuevos mecanismos de acción de los fármacos.En este estudio, identificaron cuatro proteínas reguladas al alza dentro del panel de 16 proteínas y descubrieron que dos proteínas, NUDT5 y SEPTIN8, pueden servir como nuevos objetivos para la letalidad sintética, lo que proporciona una nueva estrategia para la terapia dirigida para el cáncer de próstata.
Limitaciones del estudio
Los datos de secuenciación de RNA= se han utilizado para validar el poder predictivo del panel de 16 proteínas en las culturas occidentales en su mayor parte; la validación basada en datos proteómicos arrojará más luz sobre el poder predictivo del panel en otras poblaciones étnicas.
Aunque la eficacia de este panel de 16 proteínas para predecir la BCR se ha confirmado en una cohorte considerable, todavía se requiere una mayor validación de la cohorte clínica antes de que este modelo pueda aplicarse más en la clínica. También reconocemos que esta investigación no ha confirmado completamente la importancia funcional de estas 16 proteínas en el inicio y avance del PCa.
Ronald Palacios Castrillo