Ronald Palacios Castrillo
Resumen
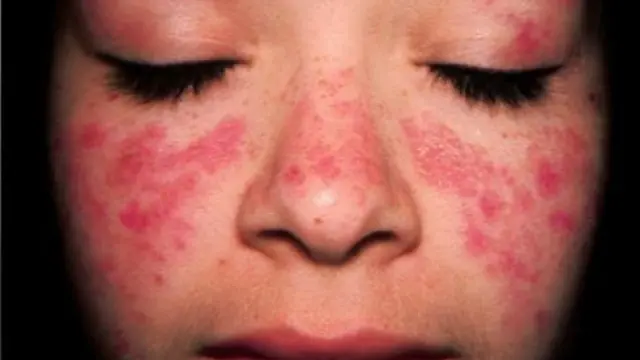
El SLE es la enfermedad autoinmune por excelencia en la que existe clara evidencia de defectos en varios genes que participan primordialmente en la identificación de células en proceso de muerte o muertas, su ingestión y ulterior degradación intracelular.La falla en la función de estos genes resulta en la acumulación de DNA y RNA en los tejidos, alcanzando niveles que estimulan una respuesta immune innata y adaptativa específica contra los ácidos nucleicos (ANA), DNA y RNA.
Se producen autoanticuerpos contra ANA, DNA y RNA que son biomarcadores del SLE y en el caso de los
autoanticuerpos contra DNAds( nativo) causantes de la patología y marcador específico del paciente con SLE.
=> Recibir por Whatsapp las noticias destacadas
Por consiguiente, situaciones que incrementen la muerte celular en los pacientes con SLE ( exposición al sol,
infecciones) pueden reactivar la enfermedad. TLR7 es un receptor transmembrana intracelular que detecta los productos de degradación del RNA dentro de los endosomas . El complejo BORC endosómico tardío junto con la pequeña GTPasa Arl8b controla los niveles intracelulares de TLR7 regulando el recambio del receptor.
Esto requiere una interacción directa entre el factor de tráfico asociado a TLR7 Unc93b1 y Arl8b. La señalización hiperactiva de TLR7 se ha considerado durante mucho tiempo como impulsora de enfermedades autoinmunes en modelos de ratón.
Recientemente, se identificaron mutaciones de ganancia de función en TLR7 como una causa monogénica ( un solo gen afectado) de SLE en humanos. En cuatro pacientes con lupus de inicio infantil, Wolf et al. identificaron variantes en la proteína chaperona transmembrana UNC93B1 que condujeron a la hiperactivación de TLR7, a la producción constitutiva de interferón tipo I y a la enfermedad SLE. Mishra, et.al.,(SCIENCE IMMUNOLOGY.Vol 9, Issue 92.DOI: 10.1126/sciimmunol.adi9575 )
Identificaron una mutación UNC93B1 en un paciente con SLE de inicio en la infancia, que da como resultado una interacción BORC reducida y una acumulación endosómica de TLR7.
Por lo tanto, la falta de control del recambio de TLR7 y su consiguiente hiperactivación con RNA dentro de las células que participan en la respuesta inmune es suficiente para romper la tolerancia inmunológica a los
ácidos nucleicos y desarrollar SLE.
Estos resultados resaltan la importancia de un sistema de endomembrana intacto en la prevención de la señalización patológica de TLR7 y las enfermedades autoinmunes como SLE.
En Detalle
Los receptores tipo Toll (TLR) sensores de ácido nucleico, incluido el sensor de RNA TLR7, son responsables de la difícil tarea de reconocer y responder eficientemente al material genómico extraño mientras se mantiene la tolerancia a los ácidos nucleicos autoderivados (1).
Esta discriminación se logra mediante la localización intracelular de TLR7 dentro de los endosomas tardíos, lo
que garantiza una detección efectiva del RNA liberado por los patógenos ingeridos pero limita la exposición de TLR7 a ligandos autoderivados.
Para llegar a los endosomas tardíos y volverse competente en señalización, TLR7 se basa en una interacción compleja entre mecanismos de transporte, interacciones de membrana y procesamiento proteolítico en
compartimentos endosómicos ácidos (2–4).
Los estudios mecanicistas sobre la ganancia de función de TLR7 se han centrado principalmente en mutaciones programadas en TLR7 o en su homólogo B1 del factor
de tráfico asociado unc-93 (Unc93b1).
Las fuentes conocidas de TLR7 hiperactivo incluyen duplicaciones del gen Tlr7 (5–7), mutaciones que aumentan la afinidad del ligando (8) o mutaciones Unc93b1 que liberan el freno de la señalización de TLR7 (9–11).
Sin embargo, no se han descrito los mecanismos reguladores de la actividad de TLR7 que involucran el orgánulo que contiene el receptor o los procesos de tráfico relacionados con el endosoma.
Los endosomas/lisosomas tardíos, desde los que emite señales TLR7, son el principal orgánulo degradativo en las células eucariotas que también sirven como coordinador central y centro de señalización de la célula
(12).
Las proteínas accesorias y transmembrana se ensamblan en complejos funcionales en la membrana endosómica para orquestar la respuesta de procesos celulares clave a señales ambientales. Uno de estos complejos es el complejo relacionado con la biogénesis del complejo 1 de orgánulos lisosomales (BLOC1)
(BORC), un regulador crítico de la biología de los lisosomas que controla el posicionamiento de los lisosomas dentro del citoplasma, así como la fusión de vesículas de endosomas tardíos y autofagosomas con lisosomas (13 ).
BORC está compuesto por ocho subunidades conservadas que se ensamblan en la superficie citosólica del endosoma (14).
El complejo recluta el pequeño factor de ribosilación 8B (Arl8b) de adenosina 5′-difosfato de guanosina trifosfatasa (GTPasa) que sirve como adaptador para conectar BORC a diferentes maquinarias de proteínas posteriores para mediar en el tráfico y la fusión de vesículas. Mishra, et.al.,(SCIENCE IMMUNOLOGY.Vol 9, Issue 92.DOI: 10.1126/sciimmunol.adi9575 ) describe que tanto BORC como Arl8b son necesarios para mantener la tolerancia inmunológica mediante la regulación del recambio homeostático de TLR7.
Por lo tanto, la clasificación degradativa mediada por BORC controla el nivel intracelular de TLR7 funcional y limita la autorreactividad a los ácidos nucleicos.
Para coordinar la degradación lisosomal de TLR7, el complejo BORC interactúa físicamente con TLR7 a través de Unc93b1.
Encontraron a un paciente con una mutación heterocigótica UNC93B1 que debilita esta interacción,
lo que lleva a la acumulación endosómica de TLR7 asociada con SLE de inicio en la infancia.
El trabajo de Mishra y colegas destaca la importancia de un sistema de endomembrana intacto para proporcionar un recambio homeostático bien controlado de TLR7 para prevenir la activación aberrante y las
enfermedades autoinmunes.
Está bien establecido que los niveles de expresión del gen Tlr7 deben controlarse estrictamente para evitar una estimulación excesiva (5–7, 35); por lo tanto, también se esperaría que los mecanismos que ralentizan la degradación de la proteína TLR7 rompieran la autotolerancia.
Identificaron el complejo BORC endosómico tardío como un componente crítico para la clasificación degradativa específica de TLR7, un proceso importante para mantener niveles adecuados de TLR7 endosómico y un umbral de activación saludable para la señalización.
La deficiencia de BORC provocó una acumulación de TLR7 endosómico en macrófagos en reposo, sensibilizando así a la célula a la activación posterior con ligandos de RNA.
La falta de efectos notables de BORC en la señalización de TLR9 podría sugerir que no todos los TLR que detectan ácidos nucleicos se degradan por la misma vía. La evidencia experimental ha demostrado que TLR7 y TLR9 toman rutas de tráfico distintas desde el retículo endoplásmico (RE) hasta los endosomas con diferentes requisitos regulatorios (36), por lo que es razonable suponer que TLR9 atraviesa diferentes compartimentos endosómicos y, por lo tanto, no está sujeto a la regulación mediada por BORC.
Los datos sugieren que Arl8b-BORC se relaciona específicamente con TLR7 y que esta interacción es necesaria para reclutar el complejo de anclaje más general HOPS para mediar en la fusión lisosomal y la renovación del receptor.
Una duplicación de E49 en UNC93B1, que identificaron en un paciente joven con lupus, debilita la interacción
entre TLR7 y el complejo BORC-ARL8B, lo que resulta en una reducción de la clasificación degradativa y la
acumulación de TLR7 en los endosomas tardíos. El alelo del paciente existe como variante monoalélica, lo que indica un efecto dominante sobre la actividad de TLR7. Sin embargo, la penetrancia de la enfermedad es
incompleta, como lo demuestra la ausencia de síntomas de la enfermedad en el padre, que también es portador.
La penetrancia incompleta de la enfermedad es común para los alelos de riesgo asociados al lupus, porque
también se sabe que contribuyen factores ambientales y (epi) genéticos (40). Sin embargo, tras una observación más cercana, el padre también reveló signos de inflamación de bajo grado evidentes mediante el inmunofenotipado de PBMC. Recientemente, se han descrito familias adicionales con mutaciones UNC93B1 en distintas regiones proteicas que también provocan la enfermedad del lupus (11). Juntos, estos dos informes establecen un papel clave para Unc93b1 en la prevención de la señalización excesiva de TLR7 y la autoinmunidad en humanos.
La participación del complejo BORC en la regulación inmune no se ha descrito previamente y podría haber
quedado oscurecida por el papel central que desempeña BORC en el mantenimiento de la integridad
neuronal.
A pesar de su expresión ubicua, el complejo BORC-Arl8b es particularmente importante en el transporte de vesículas de largo alcance dentro de los axones (41). Como resultado, los ratones con función BORC atenuada desarrollan distrofia axonal progresiva y función motora deteriorada (42).
Los ratones KO globales de subunidades BORC individuales o Arl8b no son viables (22, 42–44). Estos estudios en animales dan motivos para predecir que los humanos con función BORC atenuada presentarían principalmente trastornos neurodegenerativos, que, en particular, con frecuencia coexisten con enfermedades autoinflamatorias o autoinmunes y a menudo comparten variantes y vías genéticas (45). Esto también podría explicar por qué hasta ahora no se han asociado mutaciones BORC con enfermedades autoinmunes en estudios de asociación de todo el genoma.
El complejo BORC-Arl8b facilita múltiples funciones celulares, y los autores mostraron que no todos los tipos
de células utilizan estas funciones por igual.
En las células HeLa y otras líneas celulares epiteliales, BORC regula el posicionamiento de los lisosomas y la
autofagia (14, 20), mientras que en los macrófagos, su función principal parece ser la clasificación de la carga
endocítica y biosintética en lisosomas.
La misma función parece aplicarse a las células dendríticas, en las que la deficiencia de Arl8b retrasa la maduración lisosomal y la entrega de carga a los lisosomas, lo que resulta en una reducción de la destrucción microbiana y la presentación de antígenos (23). Los macrófagos primarios derivados del hígado fetal de ratones Borcs7 KO tampoco muestran diferencias en la posición de los lisosomas, de acuerdo con nuestros datos (42).
En las pDC estimuladas por TLR7, Arl8b controla el tráfico de vesículas que contienen TLR7 hacia la periferia celular para la inducción de IFN tipo I (46), un fenómeno que no observamos en los macrófagos, presumiblemente debido a su incapacidad general para inducir IFN tipo I en respuesta a la estimulación TLR7.
REFERENCIAS BIBLIOGRÁFICAS
1 N. A. Lind, V. E. Rael, K. Pestal, B. Liu, G. M. Barton, Regulation of the nucleic acid- sensing Toll-like receptors. Nat. Rev. Immunol. 22, 224–235 (2022).
2 S. E. Ewald, B. L. Lee, L. Lau, K. E. Wickliffe, G.-P. Shi, H. A. Chapman, G. M. Barton, The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456, 658–662 (2008).
3 S. Maschalidi, S. Hässler, F. Blanc, F. E. Sepulveda, M. Tohme, M. Chignard, P. van Endert, M. Si-Tahar, D. Descamps, B. Manoury, Asparagine endopeptidase controls anti-influenza virus immune responses through TLR7 activation. PLOS Pathog. 8, e1002841 (2012).
4 A. Kanno, C. Yamamoto, M. Onji, R. Fukui, S. I. Saitoh, Y. Motoi, T. Shibata, F. Matsumoto, T. Muta, K. Miyake, Essential role for Toll-like receptor 7 (TLR7)-unique cysteines in an intramolecular disulfide bond, proteolytic cleavage and RNA sensing. Int. Immunol. 25, 413–422 (2013).
5 J. A. Deane, P. Pisitkun, R. S. Barrett, L. Feigenbaum, T. Town, J. M. Ward, R. A. Flavell, S. Bolland, Control of Toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27, 801–810 (2007).
6 P. Pisitkun, J. A. Deane, M. J. Difilippantonio, T. Tarasenko, A. B. Satterthwaite, S. Bolland, Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672 (2006).
7 S. Subramanian, K. Tus, Q.-Z. Li, A. Wang, X.-H. Tian, J. Zhou, C. Liang, G. Bartov, L. D. McDaniel, X. J. Zhou, R. A. Schultz, E. K. Wakeland, A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U.S.A. 103, 9970–9975 (2006).
8 G. J. Brown, P. F. Cañete, H. Wang, A. Medhavy, J. Bones, J. A. Roco, Y. He, Y. Qin, J. Cappello, J. I. Ellyard, K. Bassett, Q. Shen, G. Burgio, Y. Zhang, C. Turnbull, X. Meng, P. Wu, E. Cho, L. A. Miosge, T. D. Andrews, M. A. Field, D. Tvorogov, A. F. Lopez, J. J. Babon, C. A. López, Á. Gónzalez-Murillo, D. C. Garulo, V. Pascual, T.
Levy, E. J. Mallack, D. G. Calame, T. Lotze, J. R. Lupski, H. Ding, T. R. Ullah, G. D. Walters, M. E. Koina, M. C. Cook, N. Shen, C. de Lucas Collantes, B. Corry, M. P. Gantier, V. Athanasopoulos, C. G. Vinuesa, TLR7 gain-of-function genetic variation causes human lupus. Nature 605, 349–356 (2022).
9 R. Fukui, S.-I. Saitoh, A. Kanno, M. Onji, T. Shibata, A. Ito, M. Onji, M. Matsumoto, S. Akira, N. Yoshida, K. Miyake, Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 35, 69–81 (2011).
10 O. Majer, B. Liu, L. S. M. Kreuk, N. Krogan, G. M. Barton, UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature 575, 366– 370 (2019).
11 C. Wolf, E. L. Lim, M. Mokhtari, B. Kind, A. Odainic, E. Lara-Villacanas, S. Koss, S. Mages, K. Menzel, K. Engel, G. Dückers, B. Bernbeck, D. T. Schneider, K. Siepermann, T. Niehues, C. C. Goetzke, P. Durek, K. Minden, T. Dörner, A. Stittrich, F. Szelinski, G. M. Guerra, M. Massoud, M. Bieringer, C. C. de Oliveira Mann, E. Beltrán, T. Kallinich, M.-F. Mashreghi, S. V. Schmidt, E. Latz, J. Klughammer, O. Majer, M. A. Lee-Kirsch, UNC93B1 variants underlie TLR7-dependent autoimmunity. Sci. Immunol. 9, eadi9769 (2023).
12 A. Ballabio, J. S. Bonifacino, Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118 (2020).
13 J. Pu, C. M. Guardia, T. Keren-Kaplan, J. S. Bonifacino, Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
14 J. Pu, C. Schindler, R. Jia, M. Jarnik, P. Backlund, J. S. Bonifacino, BORC, a multisubunit complex that regulates lysosome positioning. Dev. Cell 33, 176–188 (2015).
15 D. E. Johnson, P. Ostrowski, V. Jaumouillé, S. Grinstein, The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 212, 677–692 (2016).
16 J. P. Caviston, A. L. Zajac, M. Tokito, E. L. F. Holzbaur, Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol. Biol. Cell 22, 478–492 (2011).
17 C. Erie, M. Sacino, L. Houle, M. L. Lu, J. Wei, Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 42, 1941–1951 (2015).
18 D. Rigante, C. Cipolla, U. Basile, F. Gulli, M. C. Savastano, Overview of immune abnormalities in lysosomal storage disorders. Immunol. Lett. 188, 79–85 (2017).
19 T. Into, M. Inomata, E. Takayama, T. Takigawa, Autophagy in regulation of Toll-like receptor signaling. Cell. Signal. 24, 1150–1162 (2012).
20 R. Jia, C. M. Guardia, J. Pu, Y. Chen, J. S. Bonifacino, BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 13, 1648–1663 (2017).
21 A. C. Borchers, L. Langemeyer, C. Ungermann, Who’s in control? Principles of Rab GTPase activation in endolysosomal membrane trafficking and beyond. J. Cell Biol. 220, e202105120 (2021).
22 C. J. Bell, N. Gupta, K. D. Tremblay, J. Mager, Borcs6 is required for endo-lysosomal degradation during early development. Mol. Reprod. Dev. 89, 337–350 (2022).
23 S. Garg, M. Sharma, C. Ung, A. Tuli, D. C. Barral, D. L. Hava, N. Veerapen, G. S. Besra, N. Hacohen, M. B. Brenner, Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity 35, 182–193 (2011).
24 J. Anderson, G. Walker, J. Pu, BORC-ARL8-HOPS ensemble is required for lysosomal cholesterol egress through NPC2. Mol. Biol. Cell 33, ar81 (2022).
25 D. Khatter, V. B. Raina, D. Dwivedi, A. Sindhwani, S. Bahl, M. Sharma, The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci. 128, 1746–1761 (2015).
26 H. Ishida, J. Asami, Z. Zhang, T. Nishizawa, H. Shigematsu, U. Ohto, T. Shimizu, Cryo-EM structures of Toll-like receptors in complex with UNC93B1. Nat. Struct. Mol. Biol. 28, 173–180 (2021).
27 Y. M. Kim, M. M. Brinkmann, M. E. Paquet, H. L. Ploegh, UNC93B1 delivers nucleotide-sensing Toll-like receptors to endolysosomes. Nature 452, 234–238 (2008).
28 O. Majer, B. Liu, B. J. Woo, L. S. M. Kreuk, E. van Dis, G. M. Barton, Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature 575, 371–374 (2019).
29 S. G. Tangye, W. Al-Herz, A. Bousfiha, C. Cunningham-Rundles, J. L. Franco, S. M. Holland, C. Klein, T. Morio, E. Oksenhendler, C. Picard, A. Puel, J. Puck, M. R. J. Seppänen, R. Somech, H. C. Su, K. E. Sullivan, T. R. Torgerson, I. Meyts, Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 42, 1473–
1507 (2022).
30 S. A. Jenks, K. S. Cashman, E. Zumaquero, U. M. Marigorta, A. V. Patel, X. Wang, D. Tomar, M. C. Woodruff, Z. Simon, R. Bugrovsky, E. L. Blalock, C. D. Scharer, C. M. Tipton, C. Wei, S. S. Lim, M. Petri, T. B. Niewold, J. H. Anolik, G. Gibson, F. E.-H. Lee, J. M. Boss, F. E. Lund, I. Sanz, Distinct effector B cells induced by unregulated
Toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 49, 725–739.e6 (2018).
31 A. Y. Lee, H. Korner, CC chemokine receptor 6 (CCR6) in the pathogenesis of systemic lupus erythematosus. Immunol. Cell Biol. 98, 845–853 (2020).
32 T. Nogimori, Y. Sugawara, M. Higashiguchi, H. Murakami, H. Akita, S. Takahama, S. Tanaka, T. Yamamoto, OMIP 078: A 31-parameter panel for comprehensive immunophenotyping of multiple immune cells in human peripheral blood mononuclear cells. Cytometry A 99, 893–898 (2021).
33 E. Grage-Griebenow, H. D. Flad, M. Ernst, Heterogeneity of human peripheral blood monocyte subsets. J. Leukoc. Biol. 69, 11–20 (2001).
34 G. I. Rice, G. M. A. Forte, M. Szynkiewicz, D. S. Chase, A. Aeby, M. S. Abdel- Hamid, S. Ackroyd, R. Allcock, K. M. Bailey, U. Balottin, C. Barnerias, G. Bernard, C. Bodemer, M. P. Botella, C. Cereda, K. E. Chandler, L. Dabydeen, R. C. Dale, C. De Laet, C. G. E. L. De Goede, M. D. Toro, L. Effat, N. N. Enamorado, E. Fazzi, B.
Gener, M. Haldre, J.-P. S.-M. Lin, J. H. Livingston, C. M. Lourenco, W. Marques Jr., P. Oades, P. Peterson, M. Rasmussen, A. Roubertie, J. L. Schmidt, S. A. Shalev, R. Simon, R. Spiegel, K. J. Swoboda, S. A. Temtamy, G. Vassallo, C. N. Vilain, J. Vogt, V. Wermenbol, W. P. Whitehouse, D. Soler, I. Olivieri, S. Orcesi, M. S. Aglan, M. S.
Zaki, G. M. H. Abdel-Salam, A. Vanderver, K. Kisand, F. Rozenberg, P. Lebon, Y. J. Crow, Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 12, 1159–1169 (2013).
35 Z. R. Newman, J. M. Young, N. T. Ingolia, G. M. Barton, Differences in codon bias and GC content contribute to the balanced expression of TLR7 and TLR9. Proc. Natl. Acad. Sci. U.S.A. 113, E1362–E1371 (2016).
36 B. L. Lee, J. E. Moon, J. H. Shu, L. Yuan, Z. R. Newman, R. Schekman, G. M. Barton, UNC93B1 mediates differential trafficking of endosomal TLRs. eLife 2, e00291 (2013).
37 C. M. Hickey, C. Stroupe, W. Wickner, The major role of the Rab Ypt7p in vacuole fusion is supporting HOPS membrane association. J. Biol. Chem. 284, 16118–16125 (2009).
38 M. L. Jongsma, J. Bakker, B. Cabukusta, N. Liv, D. van Elsland, J. Fermie, J. L. Akkermans, C. Kuijl, S. Y. van der Zanden, L. Janssen, D. Hoogzaad, R. van der Kant, R. H. Wijdeven, J. Klumperman, I. Berlin, J. Neefjes, SKIP-HOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. EMBO J. 39, e102301 (2020).
39 T. Nishiya, E. Kajita, S. Miwa, A. L. DeFranco, TLR3 and TLR7 are targeted to the same intracellular compartments by distinct regulatory elements. J. Biol. Chem. 280, 37107–37117 (2005).
40 G. C. Tsokos, M. S. Lo, P. Costa Reis, K. E. Sullivan, New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 12, 716–730 (2016).
41 G. G. Farías, C. M. Guardia, R. De Pace, D. J. Britt, J. S. Bonifacino, BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. U.S.A. 114, E2955–E2964 (2017).
42 J. N. Snouwaert, R. J. Church, L. Jania, M. T. Nguyen, M. L. Wheeler, A. Saintsing, P. Mieczkowski, F. P. M. de Villena, D. Armao, S. S. Moy, D. N. Lorenzo, B. H. Koller, A mutation in the Borcs7 subunit of the lysosome regulatory BORC complex results in motor deficits and dystrophic axonopathy in mice. Cell Rep. 24, 1254–1265 (2018).
43 R. De Pace, D. J. Britt, J. Mercurio, A. M. Foster, L. Djavaherian, V. Hoffmann, D. Abebe, J. S. Bonifacino, Synaptic vesicle precursors and lysosomes are transported by different mechanisms in the axon of mammalian neurons. Cell Rep. 31, 107775 (2020).
44 M. Oka, K. Hashimoto, Y. Yamaguchi, S. I. Saitoh, Y. Sugiura, Y. Motoi, K. Honda, Y. Kikko, S. Ohata, M. Suematsu, M. Miura, K. Miyake, T. Katada, K. Kontani, Arl8b is required for lysosomal degradation of maternal proteins in the visceral yolk sac endoderm of mouse embryos. J. Cell Sci. 130, 3568–3577 (2017).
45 “Association between neurodegenerative diseases and autoimmune diseases.” Frontiers in Neuroscience – Research Topic (2018–2019); .
46 S.-I. Saitoh, F. Abe, A. Kanno, N. Tanimura, Y. Mori Saitoh, R. Fukui, T. Shibata, K. Sato, T. Ichinohe, M. Hayashi, K. Kubota, H. Kozuka-Hata, M. Oyama, Y. Kikko, T. Katada, K. Kontani, K. Miyake, TLR7 mediated viral recognition results in focal type I interferon secretion by dendritic cells. Nat. Commun. 8, 1592 (2017).
47 A. W. Roberts, L. M. Popov, G. Mitchell, K. L. Ching, D. J. Licht, G. Golovkine, G. M. Barton, J. S. Cox, Cas9+ conditionally-immortalized macrophages as a tool for bacterial pathogenesis and beyond. eLife 8, e45957 (2019).
48 G. G. Wang, K. R. Calvo, M. P. Pasillas, D. B. Sykes, H. Häcker, M. P. Kamps, Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 3, 287–293 (2006).
49 V. Redecke, R. Wu, J. Zhou, D. Finkelstein, V. Chaturvedi, A. A. High, H. Häcker, Hematopoietic progenitor cell lines with myeloid and lymphoid potential. Nat. Methods 10, 795–803 (2013).
50 A. E. Carpenter, T. R. Jones, M. R. Lamprecht, C. Clarke, I. Kang, O. Friman, D. A. Guertin, J. Chang, R. A. Lindquist, J. Moffat, P. Golland, D. M. Sabatini, CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 (2006).
51 J. Vandesompele, K. De Preter, F. Pattyn, B. Poppe, N. Van Roy, A. De Paepe, F. Speleman, Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1 (2002).
52 UniProt Consortium, UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
53 A. M. Waterhouse, J. B. Procter, D. M. Martin, M. Clamp, G. J. Barton, Jalview version 2–A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
54 F. Sievers, A. Wilm, D. Dineen, T. J. Gibson, K. Karplus, W. Li, R. Lopez, H. McWilliam, M. Remmert, J. Söding, J. D. Thompson, D. G. Higgins, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol. 7, 539 (2011).
55 N. A. Kulak, G. Pichler, I. Paron, N. Nagaraj, M. Mann, Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014).
56 I. Gielisch, D. Meierhofer, Metabolome and proteome profiling of complex I deficiency induced by rotenone. J. Proteome Res. 14, 224–235 (2015).
57 L. Martens, H. Hermjakob, P. Jones, M. Adamski, C. Taylor, D. States, K. Gevaert, J. Vandekerckhove, R. Apweiler, PRIDE: The proteomics identifications database. Proteomics 5, 3537–3545 (2005).
58 A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander, J. P. Mesirov, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005).
59 M. Aringer, K. Costenbader, D. Daikh, R. Brinks, M. Mosca, R. Ramsey-Goldman, J. S. Smolen, D. Wofsy, D. T. Boumpas, D. L. Kamen, D. Jayne, R. Cervera, N. Costedoat-Chalumeau, B. Diamond, D. D. Gladman, B. Hahn, F. Hiepe, S. Jacobsen, D. Khanna, K. Lerstrøm, E. Massarotti, J. McCune, G. Ruiz-Irastorza, J. Sanchez- Guerrero, M. Schneider, M. Urowitz, G. Bertsias, B. F. Hoyer, N. Leuchten, C. Tani, S. K. Tedeschi, Z. Touma, G. Schmajuk, B. Anic, F. Assan, T. M. Chan, A. E. Clarke, M. K. Crow, L. Czirják, A. Doria, W. Graninger, B. Halda-Kiss, S. Hasni, P. M. Izmirly, M. Jung, G. Kumánovics, X. Mariette, I. Padjen, J. M. Pego-Reigosa, J. Romero-Diaz, Í. Rúa-Figueroa Fernández, R. Seror, G. H. Stummvoll, Y. Tanaka, M. G. Tektonidou, C. Vasconcelos, E. M. Vital, D. J. Wallace, S. Yavuz, P. L. Meroni, M. J. Fritzler, R. Naden, T. Dörner, S. R. Johnson, 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 71, 1400–1412 (2019).
60 S. Richards, N. Aziz, S. Bale, D. Bick, S. das, J. Gastier-Foster, W. W. Grody, M. Hegde, E. Lyon, E. Spector, K. Voelkerding, H. L. Rehm, ACMG Laboratory Quality Assurance Committee, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical
Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).