La crioglobulinemia es una afección patológica caracterizada por la precipitación de inmunoglobulinas circulantes del suero humano cuando se enfría por debajo de los 4 °C; las inmunoglobulinas son reversiblemente solubles cuando se recalientan. Estas proteínas fueron descubiertas por Wintrobe y Buell en 1933. Más tarde, Lerner y Watson las identificaron como gammaglobulinas e introdujeron el término crioglobulinas (es decir, globulinas séricas precipitables en frío). Desde que surgieron las descripciones iniciales de las manifestaciones clínicas asociadas con la crioprecipitación in vitro de inmunoglobulinas a fines de la década de 1960, nuestra comprensión de la crioglobulinemia, que se caracteriza por la presencia de crioglobulinas circulantes en el suero, ha experimentado cambios considerables.
Las afecciones asociadas con este fenómeno biológico único se clasificaron en tres grupos de importancia similar: linfomas, macroglobulinemia de Waldenström y las llamadas formas esenciales, que ocurren en ausencia de cualquier enfermedad subyacente conocida.
En 1974, Brouet y sus colegas describieron un sistema de clasificación, que todavía se utiliza en la actualidad, basado en el isotipo (o isotipos) de inmunoglobulinas que constituyen la crioglobulinemia: el tipo I consiste en una inmunoglobulina monoclonal (IgM, IgG o IgA), el tipo II incluye IgG policlonal más IgM monoclonal con actividad del factor reumatoide, y el tipo III comprende IgG policlonal, IgM policlonal o ambas. Tanto el tipo II como el tipo III se conocen como crioglobulinemias mixtas.
=> Recibir por Whatsapp las noticias destacadas
Los enfoques terapéuticos empíricos han implicado diferentes combinaciones de glucocorticoides, inmunosupresores convencionales y recambio plasmático y han producido resultados inconsistentes y un mal pronóstico. La lista de causas de crioglobulinemia se ha ampliado considerablemente con el tiempo, aunque no se ha establecido la causalidad en la mayoría de los casos. A principios de la década de 1990 se produjo un cambio considerable con el descubrimiento del virus de la hepatitis C (VHC), que rápidamente se reconoció como la principal causa de lo que antes se denominaba crioglobulinemia mixta «esencial». La lista actual de causas se ha reducido a unos pocos trastornos hematológicos, enfermedades autoinmunes sistémicas e infecciones crónicas .
Los avances diagnósticos, fisiopatológicos y terapéuticos realizados desde principios de la década de 1990 han redefinido el espectro de las crioglobulinemias y han llevado a la identificación de dos afecciones distintas: la crioglobulinemia de tipo I, que se caracteriza por un trastorno genuino de la hemostasia acompañado de múltiples trombosis de vasos pequeños y medianos y, a veces, de signos de inflamación vascular; y las crioglobulinemias mixtas de tipo II y III, que se consideran un tipo de vasculitis inflamatoria genuina de vasos pequeños causada por el depósito de complejos inmunitarios mediado por el complemento.
A pesar de que ahora se reconoce mejor la crioglobulinemia que en el pasado, todavía son escasos los datos epidemiológicos sólidos y completos, y la enfermedad se considera rara. Sin embargo, una mejor comprensión de los mecanismos subyacentes condujo al desarrollo de terapias más dirigidas (contra el VHC o los clones de linfocitos) que están cambiando la historia natural de la crioglobulinemia. En vista de sus propiedades biológicas distintivas (y, por lo tanto, sus diferentes implicaciones clínicas y terapéuticas), la crioglobulinemia de tipo I y la crioglobulinemia mixta constituyen dos enfermedades distintas categorizadas bajo el mismo nombre. Esta revisión de Cacoub,et.al.,[N Engl J Med 2024;391:1426-1439.
DOI: 10.1056/NEJMra2400092] ,profundiza en un análisis detallado y una comparación de estas afecciones.
Síndrome de crioglobulinemia
Características clínicas
Las manifestaciones clínicas asociadas con el síndrome de crioglobulinemia son muy diversas y pueden afectar virtualmente a cualquier órgano; por lo tanto, el síndrome se clasifica como una enfermedad sistémica genuina. Sin embargo, no todas las manifestaciones son comunes a ambos tipos de crioglobulinemia, y ciertas diferencias sutiles pueden ser útiles para que los médicos diferencien entre la crioglobulinemia de tipo I y la crioglobulinemia mixta. Los datos que caracterizan a los pacientes con crioglobulinemia de tipo I se han derivado de cohortes retrospectivas que agruparon casos que involucraban IgG e IgM, mientras que los datos sobre pacientes con crioglobulinemia mixta provienen principalmente de casos de vasculitis relacionada con el VHC; ambos tipos se resumen en la Figura 1.
Puntos clave
Crioglobulinemia
- Las causas de las crioglobulinemias se limitan actualmente a unos pocos trastornos hematológicos, enfermedades autoinmunes sistémicas e infecciones crónicas.
- El síndrome clínico de crioglobulinemia comprende dos fenotipos principales.
- La crioglobulinemia de tipo I es un trastorno genuino de la hemostasia que conduce a la obstrucción mecánica de múltiples vasos pequeños y medianos (hiperviscosidad, trombosis o ambas); los pacientes con esta afección a veces muestran signos de inflamación vascular.
- Las crioglobulinemias mixtas de tipo II y III se caracterizan por una vasculitis autoinmune de vasos pequeños causada por el depósito de complejos inmunes mediado por el complemento.
- Mientras que la crioglobulinemia de tipo I generalmente surge de un cáncer hematológico, la crioglobulinemia mixta se caracteriza por una linfoproliferación indolente de células B que eventualmente puede conducir a una transformación manifiesta de linfoma.
- Cuando está indicada la terapia farmacológica, primero se debe identificar la enfermedad subyacente y luego se puede apuntar al clon de linaje de células B.
La afectación de la piel está presente en la mayoría de los casos de crioglobulinemia mixta y generalmente se manifiesta como púrpura vascular con lesiones que a veces son necróticas (Figura 2A). En la crioglobulinemia de tipo I, las manifestaciones cutáneas dependen en gran medida de la temperatura ambiente y la necrosis cutánea es mucho más marcada que en la crioglobulinemia mixta. La afectación articular y muscular está presente en la mayoría de los pacientes con crioglobulinemia mixta y consiste en artralgias inflamatorias, bilaterales, simétricas y no destructivas que afectan a las articulaciones grandes; la sinovitis es rara.
Las manifestaciones clínicas de la crioglobulinemia mixta pueden parecerse a las de la artritis reumatoide, en particular en presencia del factor reumatoide. Muchos pacientes son tratados en consecuencia y, cuando presentan manifestaciones sistémicas, a menudo terminan con un diagnóstico erróneo de vasculitis reumatoide. La ausencia de erosiones óseas y anticuerpos antiproteína citrulinada ayuda a distinguir entre artritis reumatoide y crioglobulinemia.
En pacientes con crioglobulinemia de tipo I, las artralgias ocurren con menos frecuencia, con hinchazón dolorosa de los dedos de las manos y de los pies que no es necesariamente de origen articular. En casos raros, la neuropatía de fibras pequeñas podría ser la causa de dolor generalizado en pacientes con crioglobulinemia.
El deterioro neurológico más frecuente en la crioglobulinemia mixta es la polineuropatía sensitivomotora. La afectación cerebral es muy rara y se manifiesta como déficits focales o trastornos neurocognitivos. Los síntomas neurológicos asociados con la infiltración de linfoma o el síndrome de hiperviscosidad también pueden contribuir a la manifestación clínica de la crioglobulinemia. Los síntomas de hiperviscosidad se producen cuando la inmunoglobulina monoclonal está presente en grandes cantidades, lo que es característico de la crioglobulinemia de tipo I, y pueden incluir sangrado de las mucosas y signos neurosensoriales.
Aproximadamente un tercio de los pacientes con crioglobulinemia mixta tienen afectación renal, generalmente en forma de glomerulonefritis membranoproliferativa. Las recaídas sucesivas pueden provocar diversos grados de fibrosis renal, aunque la insuficiencia renal terminal es, afortunadamente, poco frecuente. Las complicaciones renales también se producen en pacientes con crioglobulinemia de tipo I y pueden estar relacionadas con el cáncer hematológico subyacente. Los síntomas generales, como la fatiga y la fiebre intermitente, se asocian con frecuencia a las recaídas de la enfermedad. Pueden presentarse otros síntomas graves relacionados con vasculitis digestiva, cardíaca, pulmonar o retiniana, pero son poco frecuentes.
Los criterios de clasificación de la vasculitis crioglobulinémica mixta incluyen los aspectos más relevantes de la enfermedad. Aunque la detección de crioglobulinemia es una condición sine qua non para clasificar la vasculitis, estos criterios de clasificación pueden ser útiles para diagnosticar la crioglobulinemia en pacientes sin crioglobulinemia detectada en el suero.
Diagnóstico
La manifestación clínica del síndrome de crioglobulinemia debe confirmarse. Sin embargo, la detección de crioglobulinemia puede ser un desafío debido a los requisitos muy estrictos de muestreo. En una serie grande, el 9% de los pacientes con crioglobulinemia inicialmente tuvieron pruebas falsas negativas. Para prevenir la crioprecipitación ex vivo, las condiciones preanalíticas deben ser rigurosas: la muestra de sangre debe mantenerse a 37 °C hasta la centrifugación en el laboratorio. El crioprecipitado se manifiesta como un borde más o menos grueso en el pellet del tubo, y su cantidad se mide en gramos por litro o de forma semicuantitativa (Figura 2B). La inmunofenotipificación identifica la inmunoglobulina constituyente que determina el isotipo (tipo I, II o III en aproximadamente el 10%, 50% y 40% de los casos, respectivamente), que luego guía la evaluación etiológica. La presencia de criofibrinógeno, otra crioproteína, en el plasma puede ser un factor de confusión potencial durante las pruebas diagnósticas de crioglobulinemia tipo I.
En caso de una fuerte sospecha clínica de crioglobulinemia pero dificultad para identificar crioglobulinas circulantes, su presencia puede sospecharse sobre la base de hipergammaglobulinemia (posiblemente con un pico monoclonal en la inmunofijación de proteínas séricas), consumo de complemento (niveles bajos de C4 y C3) y factor reumatoide positivo. Además, la presencia de crioglobulinas séricas puede interferir con los análisis biológicos automatizados, lo que resulta en pseudoleucocitosis, pseudomacrocitosis e incluso hipogammaglobulinemia paradójica (debido a una importante precipitación de complejos inmunes). Se deben realizar pruebas adicionales sobre la base de la evaluación diagnóstica subyacente . En los casos en los que el diagnóstico biológico sigue siendo difícil de alcanzar, una biopsia del tejido objetivo (como la piel, el riñón o el nervio periférico) puede proporcionar evidencia concluyente (Figura 2C y 2D).
Historia natural
Gran parte de nuestro conocimiento sobre la crioglobulinemia mixta proviene de su asociación con el VHC. Entre los pacientes con crioglobulinemia mixta relacionada con el VHC, entre el 25 y el 30 % permanecen asintomáticos, entre el 40 y el 45 % tienen manifestaciones cutáneas predominantemente leves, entre el 20 y el 30 % presentan daño orgánico significativo, entre el 7 y el 12 % presentan progresión a cáncer de células B y entre el 2 y el 5 % tendrán vasculitis de progresión rápida y potencialmente mortal. En la práctica, los pacientes con crioglobulinemia de tipo III suelen tener características clínicas más leves que aquellos con enfermedad de tipo II.
En un estudio previo, después de la introducción de terapias anti-VHC basadas en interferón, que llevaron a varias respuestas virológicas sostenidas, la supervivencia general a 5 años entre los pacientes con crioglobulinemia mixta se estimó en un 75 %. Las infecciones bacterianas y la enfermedad hepática terminal fueron las principales causas de muerte, y la fibrosis hepática y la gravedad de la vasculitis (es decir, compromiso renal) fueron los principales factores pronósticos.
La llegada de la terapia con agentes antivirales de acción directa en la última década, que ha dado como resultado una respuesta virológica sostenida en más del 95% de los pacientes y que tiene un excelente perfil de seguridad, ha transformado el tratamiento y el pronóstico de la crioglobulinemia mixta relacionada con el VHC. En un estudio prospectivo de gran tamaño que incluyó a 148 pacientes con vasculitis crioglobulinémica relacionada con el VHC, solo el 2,8% murió después de un seguimiento medio de 15,3 meses.
Un estudio prospectivo que incluyó a una cohorte italiana examinó la vasculitis crioglobulinémica no relacionada con el VHC (incluida la enfermedad relacionada con el virus de la hepatitis B [VHB], las enfermedades autoinmunes subyacentes y las formas esenciales) y mostró que los pacientes varones con púrpura, los pacientes con crioglobulinemia de tipo II y los pacientes con VHB subyacente tuvieron los peores resultados. El pronóstico a largo plazo de la crioglobulinemia mixta pura no infecciosa aún está por determinar.
En un estudio de cohorte de gran tamaño que incluyó a pacientes con crioglobulinemia de tipo I, la supervivencia estimada sin recaída clínica, complicaciones del tratamiento o muerte fue del 26% a los 5 años. Los factores independientes asociados con una supervivencia libre de eventos deficiente incluyeron el isotipo de IgG y la afectación renal.
Fisiopatología
Causas de la crioglobulinemia: un concepto en evolución
Más de 55 años después de la descripción inicial de Meltzer et al., una serie de mecanismos fisiopatológicos bien descifrados nos permiten ahora delinear, dentro del término general “crioglobulinemia”, dos enfermedades distintas: la crioglobulinemia de tipo I, un proceso hematológico que implica trombosis en arterias de tamaño pequeño y mediano; y las crioglobulinemias mixtas de tipo II y III, una vasculitis autoinmune. Los mecanismos generales implicados en la crioglobulinemia se describen en la Figura 3.
A lo largo de la historia de la investigación sobre la crioglobulinemia, las formas esenciales de crioglobulinemia mixta se han vuelto cada vez más raras. El perfil demográfico de los pacientes depende en gran medida de la enfermedad subyacente. Además, las terapias antivirales directas contra el VHC han provocado un cambio considerable en el perfil epidemiológico de la crioglobulinemia mixta. Aunque las enfermedades autoinmunes (como el síndrome de Sjögren y el lupus eritematoso sistémico) representaron aproximadamente el 10 al 20% de las crioglobulinemias de tipo II y III en series más antiguas, desde 2015 han representado más del 50% de los casos, surgiendo como la principal causa de crioglobulinemia mixta. Con la disminución de la prevalencia mundial de la infección por VHC, la incidencia general de crioglobulinemia mixta ha disminuido, siendo más prevalente una edad media más joven y una mayor proporción de pacientes femeninas.
La crioglobulinemia de tipo I generalmente surge de la expansión de un solo clon dentro de las células sanguíneas patológicas, que puede ser indolente, latente o maligna. En una cohorte más reciente de pacientes con crioglobulinemia tipo I, la distribución de las condiciones hematológicas subyacentes fue la siguiente: gammapatías monoclonales de importancia clínica en el 31% de los pacientes, enfermedad de Waldenström en el 27%, linfoma no Hodgkin en el 20%, mieloma múltiple en el 15% y leucemia linfocítica crónica en el 7%.
Mecanismos de la vasculitis crioglobulinémica mixta
Como virus hepatotrópico y linfotrópico, el VHC es capaz de inducir una variedad de trastornos linfoproliferativos de células B, desde hipergammaglobulinemia policlonal aislada con crioglobulinas detectables en el suero hasta vasculitis crioglobulinémica y, en última instancia, cáncer de células B. Este espectro está respaldado por datos sólidos y ha sido ampliamente revisado en otros estudios.
La estimulación crónica del antígeno viral desempeña un papel central en la linfoproliferación relacionada con el VHC. La unión de la proteína E2 del VHC al complejo de membrana CD19–CD21–CD81 en las células B reduce el umbral de activación y proliferación del VHC e induce una hipermutación somática sesgada de las inmunoglobulinas (en el gen que codifica IgH VH1-69), lo que da lugar a hipergammaglobulinemia. Posteriormente, las crioglobulinas se producen de forma policlonal (crioglobulinemia de tipo III) y, si persiste la estimulación viral, se añade un componente monoclonal (crioglobulinemia de tipo II), lo que marca una progresión hacia la linfoproliferación autónoma. Las moléculas de IgM con actividad del factor reumatoide pueden formar complejos inmunes con IgG y fracciones del complemento, lo que conduce a la inflamación y lesión tisular.
Las células B experimentan una expansión clonal, asumiendo un fenotipo de memoria atípico (CD21−CD27+IgM+T-bet+). La expresión disminuida de CD21 (es decir, el receptor de complemento CR2) hace que las células B sean funcionalmente anérgicas a estímulos genéricos y menos propensas a ser eliminadas de la circulación. Sin embargo, las células B de memoria atípicas responden a la estimulación del receptor tipo Toll 9 e inducen una respuesta de células T auxiliares de tipo 1 (Th1) de linfocitos, a expensas de las células T reguladoras.
La expansión clonal impulsada por autoantígenos y el agotamiento de células B productoras de factor reumatoide seleccionadas ocurre en la crioglobulinemia relacionada con el VHC, formas esenciales de crioglobulinemia y crioglobulinemias resultantes del síndrome de Sjögren primario, lo que sugiere mecanismos patogénicos comunes en todas las crioglobulinemias mixtas. El estimulador de linfocitos B desempeña un papel crítico en la supervivencia de las células B, y su sobreexpresión induce la expansión de clones de células B, posiblemente contribuyendo a la progresión de la linfoproliferación observada en la crioglobulinemia relacionada con el VHC y Síndrome de Sjögren.
Con el tiempo, la proliferación inicial de células B impulsada por el VHC se vuelve gradualmente independiente del virus. La linfoproliferación insensible a los antígenos puede conducir a una transformación linfomatosa manifiesta, completando así el continuo. La vasculitis crioglobulinémica es, con diferencia, el factor de riesgo más importante de cáncer de células B en pacientes con infección crónica por el VHC, lo que confiere un riesgo 35 veces mayor en estos pacientes que en la población general. Los cánceres hematológicos relacionados con el VHC más frecuentes en este contexto son el linfoma de la zona marginal, el linfoma linfoplasmocítico y el linfoma difuso de células B grandes de alto grado.
Mecanismos de la crioglobulinemia de tipo I
La vasculopatía crioglobulinémica de tipo I tiene más similitudes con los trastornos asociados a las células plasmáticas que con las enfermedades autoinmunes. Cuando no se asocia con un trastorno linfoproliferativo manifiesto, la crioglobulinemia de tipo I se considera una gammapatía monoclonal de importancia clínica. Las crioglobulinas IgG monoclonales tienen características morfológicas finamente estructuradas y se ha demostrado que forman redes macromoleculares altamente estructuradas capaces de atrapar físicamente las células, un proceso conocido como formación de rouleaux dentro de los vasos sanguíneos.
En general, los clones de células B tienen una firma genética y molecular similar a la de la crioglobulinemia de tipo I, pero las gammapatías monoclonales y los cánceres hematológicos manifiestos forman dos extremos de un continuo en términos de la cantidad de mutaciones somáticas.
Debido a que las crioglobulinas de tipo I tienen actividad del factor reumatoide solo en casos raros, la vasculitis inflamatoria mediada por complemento generalmente ocurre con menos frecuencia con la crioglobulinemia de tipo I que con la crioglobulinemia mixta. Un modelo murino de enfermedad renal crioglobulinémica destacó el papel principal de la deposición de complejos inmunes en los capilares glomerulares en lugar de los mecanismos nefrotóxicos dependientes del complemento y del receptor Fc. Sobre todo, la obstrucción mecánica de la microcirculación causada por microtrombosis es el mecanismo primario subyacente a la crioglobulinemia de tipo I.
Enfoques terapéuticos
En las últimas dos décadas, las estrategias terapéuticas para la crioglobulinemia se han vuelto cada vez más específicas debido a los avances en nuestra comprensión de los mecanismos subyacentes de la enfermedad. El tratamiento de la crioglobulinemia mixta a menudo implica agentes antivirales de acción directa contra el VHC, con o sin rituximab (un anticuerpo monoclonal anti-CD20), y con mucha menos frecuencia implica el uso de glucocorticoides. Para la crioglobulinemia de tipo I, los regímenes de tratamiento pueden incluir glucocorticoides, intercambio de plasma e inmunosupresores. La plasmaféresis es una opción terapéutica en el contexto de la enfermedad por IgM porque el 80% de los anticuerpos IgM permanecen en la circulación. El abordaje terapéutico de las crioglobulinemias se muestra en la Figura 4.
Crioglobulinemias mixtas de tipo II y III
En los casos asociados con el VHC, la erradicación del virus es imperativa. Aunque la linfoproliferación de células B es impulsada predominantemente por estímulos virales crónicos, una respuesta virológica sostenida debería ser suficiente para eliminarla. Se ha demostrado que los agentes antivirales de acción directa por sí solos reducen la actividad de la vasculitis crioglobulinémica y restablecen el equilibrio entre el aumento de las células B de memoria atípicas y los linfocitos T patógenos y la disminución de las células T reguladoras. La presencia persistente de crioglobulinas en el suero puede servir como un marcador sustituto de la linfoproliferación indolente. Un estudio de cohorte internacional muy grande mostró que hasta el 12,6% de los pacientes tienen una recaída de la vasculitis a largo plazo a pesar de una respuesta virológica sostenida adecuada después de la terapia con un agente antiviral de acción directa, similar a los porcentajes observados en un estudio italiano multicéntrico. Además de las crioglobulinas persistentes, la linfocitosis de células B monoclonales combinadas, la translocación t(14;18) y las proporciones anormales de cadena ligera libre kappa a lambda se han sugerido recientemente como predictores de la respuesta clínica después de una respuesta virológica sostenida. La linfoproliferación insensible al antígeno y la proliferación descontrolada de células B pueden dar lugar a una progresión hacia un linfoma manifiesto que justifica un tratamiento específico dirigido al linfoma.
En general, los regímenes terapéuticos están determinados por la gravedad de la vasculitis e incluyen la erradicación de los desencadenantes virales en los casos con causas infecciosas. En pacientes con enfermedad leve a moderada (p. ej., artralgias, mialgias, púrpura no necrótica o neuropatía puramente sensorial), la terapia antiviral de acción directa sola suele ser suficiente para lograr una respuesta virológica sostenida y la remisión de la vasculitis. En pacientes con formas graves (p. ej., glomerulonefritis con deterioro de la función renal, déficits motores, mononeuropatía múltiple, necrosis cutánea extensa o compromiso digestivo, cardíaco o pulmonar), la terapia antiviral de acción directa debe combinarse con rituximab (375 mg por metro cuadrado de superficie corporal los días 1, 8, 15 y 22 de tratamiento). En caso de manifestaciones potencialmente fatales, se pueden agregar plasmaféresis y pulsos de glucocorticoides. En pacientes que no han tenido respuesta a los agentes antivirales de acción directa o que tienen contraindicaciones para estos agentes, se puede utilizar rituximab solo como tratamiento de inducción, seguido de una infusión de 500 mg cada 6 a 9 meses como tratamiento de mantenimiento.
Varios ensayos clínicos y estudios observacionales mostraron una buena eficacia y un perfil de seguridad favorable de rituximab en pacientes con crioglobulinemia mixta grave.
Sin embargo, los médicos deben tener en cuenta un evento adverso particular que parece ser muy específico de la crioglobulinemia: la aparición de un brote de vasculitis inducido por fármacos, similar a la enfermedad del suero, entre los días 2 y 9 después de la infusión de rituximab, con una mortalidad asociada que excede el 50%. Entre los factores de riesgo de esta grave complicación se encuentran el aumento de los niveles de crioglobulina, el aumento de la gravedad de la enfermedad y dos dosis de 1000 mg de rituximab administradas con 2 semanas de diferencia. Por esta razón, recomendamos cuatro infusiones semanales de 375 mg como régimen de inducción, junto con la premedicación estándar con metilprednisolona, paracetamol y clorfenamina antes de cada infusión. Si ocurre una recaída clínica después de la interrupción del rituximab, el retratamiento con rituximab solo parece ser una estrategia efectiva y segura a largo plazo.
La interleucina-2 en dosis bajas está surgiendo como un tratamiento potencial en distintos escenarios que involucran enfermedad autoinmune y podría servir como una alternativa para casos refractarios. Se investigó en un estudio prospectivo de fase 1-2a, abierto, en el que se demostró que tiene un perfil de seguridad favorable y condujo a una mejoría clínica en 8 de 10 pacientes, además de la restauración del equilibrio entre las células T reguladoras y efectoras.
Para los casos raros de vasculitis crioglobulinémica debido a la infección por VHB o la infección por el virus de la inmunodeficiencia humana, apuntar a la estimulación viral crónica también debería ser un objetivo primario. Aunque el enfoque terapéutico general utilizado en casos de replicación del VHB es similar al utilizado para la infección por VHC, el rituximab debe utilizarse con precaución en casos de replicación del VHB. En el caso de la crioglobulinemia mixta no infecciosa, el tratamiento se adapta a la causa fisiopatológica de la enfermedad subyacente, que generalmente es una enfermedad autoinmune con actividad excesiva de células B (p. ej., lupus eritematoso sistémico o síndrome de Sjögren).
Los glucocorticoides, con o sin metotrexato, generalmente son suficientes para las formas cutáneo-articulares, mientras que la colchicina puede prescribirse para la vasculitis cutánea aislada. En casos graves de crioglobulinemia mixta, se recomienda el rituximab, en particular en presencia de compromiso renal o neurológico o para formas cutáneo-articulares refractarias. La ciclofosfamida se utiliza en casos refractarios y los intercambios plasmáticos pueden utilizarse en casos que involucran manifestaciones graves o potencialmente mortales.
Crioglobulinemia tipo I
En pacientes con indicación de tratamiento sistémico (generalmente debido a ulceración cutánea), es fundamental apuntar al clon de células plasmáticas o al clon de células linfoplasmocíticas, ya que cualquiera de ellos acabará provocando otras manifestaciones hematológicas graves. Dada la dificultad de realizar ensayos clínicos prospectivos en este contexto tan poco frecuente, en los últimos años se han logrado pocos avances. El pronóstico de la crioglobulinemia tipo I no ha mejorado con el tiempo, con una supervivencia global a 5 años que oscila entre el 77 y el 83% en estudios que incluyeron pacientes reclutados entre 1983 y 2018. El tratamiento del trastorno monoclonal subyacente en pacientes con crioglobulinemia tipo I da como resultado la eliminación de crioglobulinas séricas en solo la mitad de los pacientes, a pesar de una disminución de los síntomas.
Actualmente, no se ha establecido un estándar de atención o pautas internacionales para el tratamiento de la crioglobulinemia tipo I, y las recomendaciones se derivan principalmente de la opinión de expertos. Las opciones terapéuticas disponibles son bastante diversas porque las estrategias actuales se basan principalmente en medicamentos específicos que abordan la afección hematológica subyacente.
En los casos en los que no hay una indicación hematológica clara, la elección del tratamiento para la crioglobulinemia tipo I generalmente se guía por el isotipo de inmunoglobulina monoclonal. Para el isotipo IgM, se puede utilizar rituximab, a veces combinado con bendamustina o ciclofosfamida. En el caso del isotipo IgG o IgA, las combinaciones de tratamiento pueden incluir bortezomib, agentes anti-CD38 (p. ej., daratumumab o isatuximab) u otros fármacos que tengan como objetivo las células plasmáticas. Los intercambios plasmáticos se utilizan habitualmente. También se pueden añadir glucocorticoides e inmunosupresores en diversos grados como parte del régimen de tratamiento. Sin embargo, algunos pacientes que presentan solo púrpura pueden ser tratados con medidas no farmacológicas, como medias de compresión. Evitar la exposición al frío es importante para todos los pacientes que presentan crioglobulinemia tipo I.
Perspectivas futuras
Aunque el tratamiento con rituximab produce una respuesta clínica satisfactoria en pacientes con crioglobulinemia mixta, es posible que no restaure eficazmente los puntos de control de tolerancia de las células B tempranas que están defectuosos. Las concentraciones séricas elevadas del estimulador de linfocitos B en pacientes con crioglobulinemia mixta se asocian con un aumento de la proliferación de células B, de los niveles séricos de crioglobulina y de la actividad de la vasculitis y, por lo tanto, pueden contribuir a la recaída. A la luz de las observaciones retrospectivas en pacientes con crioglobulinemia mixta que tenían una enfermedad refractaria al rituximab solo, los ensayos clínicos en curso que involucran a pacientes con crioglobulinemia mixta no infecciosa están evaluando la combinación terapéutica secuencial de rituximab y belimumab (un anticuerpo monoclonal antiestimulador de linfocitos B; número ClinicalTrials.gov, NCT04629144), así como investigando la eficacia y la seguridad del agente anti-CD20 de próxima generación obinutuzumab. En pacientes con crioglobulinemia tipo I, el uso de agentes alquilantes y bortezomib se asocia con varios efectos secundarios.
A pesar de su importancia en los mecanismos fisiopatológicos subyacentes de la enfermedad, las células plasmáticas autorreactivas no han sido sistemáticamente atacadas en la crioglobulinemia tipo I. La inhibición del CD38 ha tenido éxito en el tratamiento de trastornos refractarios de células plasmáticas como el mieloma múltiple o la amiloidosis AL. La inhibición del CD38 en pacientes con crioglobulinemia tipo I es prometedora, y un estudio piloto abierto de fase 2 en curso que evalúa la eficacia y la seguridad del isatuximab (un anticuerpo monoclonal anti-CD38; número ClinicalTrials.gov, NCT05114109) puede proporcionar más información sobre el tratamiento de estos pacientes. Finalmente, otros métodos prometedores (por ejemplo, la terapia que implica tecnología de receptores de antígenos quiméricos o anticuerpos biespecíficos) se están expandiendo rápidamente en el manejo de afecciones hematológicas y de mediación inmunológica y podrían representar nuevas opciones en el tratamiento de la crioglobulinemia.
Conclusión
Más de medio siglo después de su descripción inicial, las crioglobulinemias mixtas y de tipo I deben considerarse como dos entidades distintas, cada una caracterizada por mecanismos subyacentes, enfoques terapéuticos y pronósticos únicos. Los avances fisiopatológicos y terapéuticos en la crioglobulinemia relacionada con el VHC durante las últimas tres décadas han sido considerables y han llevado a mejoras sustanciales en la atención al paciente. El mayor énfasis en las enfermedades autoinmunes subyacentes ha subrayado la brecha de conocimiento existente en este dominio. La variedad de afecciones hematológicas subyacentes en pacientes con crioglobulinemia de tipo I resalta los desafíos inherentes al estudio de esta afección. Los esfuerzos futuros deben concentrarse en la investigación traslacional y los ensayos aleatorizados multicéntricos centrados en la crioglobulinemia mixta no infecciosa y la crioglobulinemia de tipo I, con el objetivo final de mejorar el pronóstico.
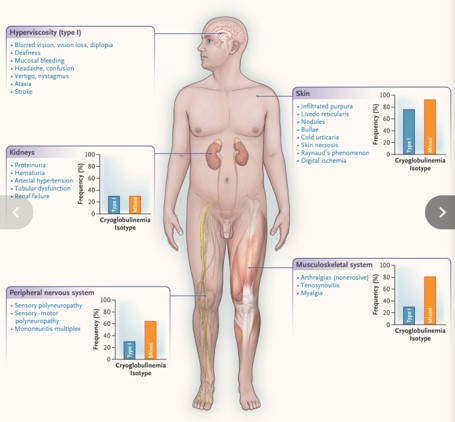
Figura 1
Presentación clínica del síndrome de crioglobulinemia.
Se muestran las manifestaciones clínicas más frecuentes del síndrome de crioglobulinemia, junto con la frecuencia de cada categoría de características clínicas según el isotipo de crioglobulinemia. La afectación cutánea incluye púrpura vascular, que generalmente depende de la gravedad y que comienza en las extremidades inferiores y se extiende a las extremidades superiores, evitando el tronco y la cara. Los brotes recurrentes pueden dejar un rastro de dermatitis con un color ocre. La afectación cutánea también puede incluir livedo reticularis, nódulos subcutáneos, ampollas, vesículas, urticaria por frío y parestesia en los dedos de las manos y los pies, en lugar del verdadero fenómeno de Raynaud.
Estas características de la afectación cutánea están estrechamente relacionadas con el esfuerzo físico y el ortostatismo en las crioglobulinemias mixtas de tipo II y III, mientras que el papel del frío externo es sustancial en la crioglobulinemia de tipo I. Las manifestaciones cutáneas observadas en la crioglobulinemia de tipo I implican principalmente hipoperfusión de las extremidades (p. ej., necrosis acral o fenómeno de Raynaud), mientras que la crioglobulinemia mixta se manifiesta predominantemente como lesiones inflamatorias (p. ej., púrpura vasculítica o nódulos subcutáneos).
La afectación renal suele ser indistinguible clínicamente entre la crioglobulinemia de tipo I y la crioglobulinemia mixta y puede manifestarse como proteinuria en rango nefrótico, hipertensión arterial, hematuria, insuficiencia renal o una combinación de estas características. La afectación renal en la crioglobulinemia de tipo I puede estar relacionada con la afección hematológica subyacente (p. ej., enfermedad renal relacionada con el mieloma con o sin disfunción tubular). El deterioro neurológico más común en pacientes con crioglobulinemia mixta es la polineuropatía sensitivo-motora, que comienza en los pies y rara vez se extiende por encima de las rodillas; las manos también pueden estar afectadas, aunque la afectación generalmente no afecta las extremidades superiores por encima de las muñecas.
Los síntomas sensoriales son típicamente la característica inicial y pueden durar varios meses o incluso años antes de la aparición de los síntomas de déficit motor. Con menor frecuencia, la aparición repentina de múltiples mononeuropatías puede imitar la periarteritis nodosa. La afectación cerebral es muy rara y puede manifestarse como déficits focales o trastornos neurocognitivos. En la crioglobulinemia de tipo I, la presencia de inmunoglobulinas monoclonales en grandes cantidades puede provocar síntomas de hiperviscosidad, incluyendo sangrado de las mucosas y signos neurosensoriales. El amplio espectro de manifestaciones clínicas abarca visión borrosa o disminuida, diplopía, dolor de cabeza, confusión, sordera, vértigo, nistagmo, ataxia, accidente cerebrovascular o incluso coma.
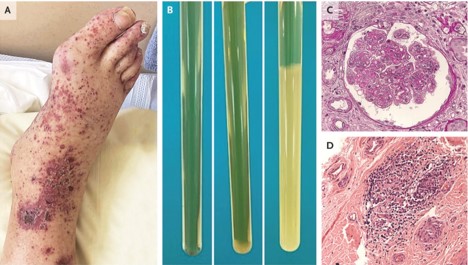
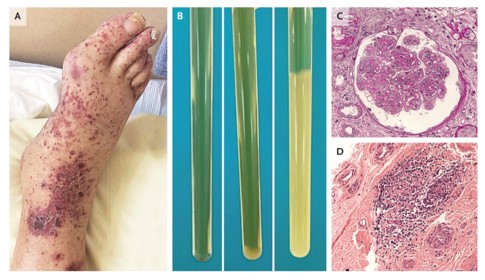
Figura 2
Diagnóstico de la crioglobulinemia.
El diagnóstico de crioglobulinemia se sospecha sobre la base de características clínicas características, como la púrpura vascular de las extremidades inferiores (Panel A). El diagnóstico se confirma mediante la detección de precipitados inducidos por el frío en las pruebas de laboratorio (Panel B). Después de la centrifugación de la sangre a 37 °C, el suero se almacena a 4 °C durante 7 días y luego se centrifuga a 4 °C: las pruebas pueden ser negativas (Panel B, izquierda), positivas con un criocrito bajo (Panel B, medio) o positivas con un criocrito muy alto (nivel sérico de crioglobulina) (Panel B, derecha).
En los casos en los que no es posible un diagnóstico biológico, las biopsias de tejido específico pueden ser útiles. La biopsia renal generalmente identifica lesiones compatibles con glomerulonefritis membranoproliferativa tipo I, que se caracteriza por el depósito de inmunoglobulina (el mismo isotipo que la crioglobulinemia) y el depósito de complemento (C3 y C1q) en la inmunofluorescencia. La proliferación endocapilar, la vasculitis o la trombosis de las arterias intrarrenales pequeñas son comunes.
Se muestra una muestra de biopsia renal con un glomérulo agrandado con duplicación de la membrana basal glomerular, hipercelularidad mesangial, hipercelularidad endocapilar y positividad para pseudotrombos en la tinción de ácido peryódico-Schiff (Panel C). La microscopía electrónica mostraría un patrón de doble contorno de la membrana basal glomerular. También se puede realizar una biopsia de nervio periférico que, por lo general, mostraría un infiltrado linfocítico perivascular adyacente a los nervios afectados (Panel D).
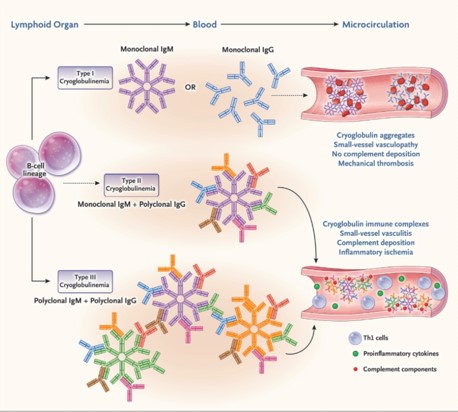
Figura 3
Mecanismos de la crioglobulinemia.
La crioglobulinemia tiene dos mecanismos subyacentes distintos: la crioglobulinemia de tipo I evoluciona como una vasculopatía que afecta a las arterias de tamaño pequeño y mediano, mientras que las crioglobulinemias mixtas de tipo II y III se manifiestan como una vasculitis autoinmune verdadera que afecta a los vasos de tamaño pequeño a mediano. En ambos tipos, los linfocitos del linaje de células B (como las células B de memoria o las células plasmáticas) producen crioglobulinas de manera monoclonal o policlonal. Las crioglobulinas de tipo I son IgM o IgG monoclonales producidas en grandes cantidades por las células plasmáticas, formando redes macromoleculares muy compactas que atrapan físicamente las células dentro de los vasos sanguíneos, un proceso conocido como formación de rouleaux.
Debido a que las crioglobulinas de tipo I rara vez tienen actividad del factor reumatoide, la vasculitis inflamatoria mediada por el complemento es poco frecuente. En cambio, el mecanismo principal en la crioglobulinemia de tipo I implica la obstrucción vascular mecánica por agregados inducidos por el frío dentro de la microcirculación, lo que conduce a microtrombosis de vasos pequeños. Las crioglobulinas mixtas consisten exclusivamente en inmunoglobulinas policlonales (tipo III) o una combinación de inmunoglobulinas policlonales con IgM monoclonal (tipo II). Esta IgM típicamente tiene actividad de factor reumatoide, que activa el complemento dentro de los complejos inmunes, lo que finalmente conduce a la inflamación y lesión tisular. Otro mecanismo clave en la crioglobulinemia mixta es la expansión e infiltración tisular de células T auxiliares de linfocitos tipo 1 (Th1) y células T efectoras, que producen citocinas proinflamatorias como el factor de necrosis tumoral α y el interferón-γ, a expensas de las células T reguladoras.
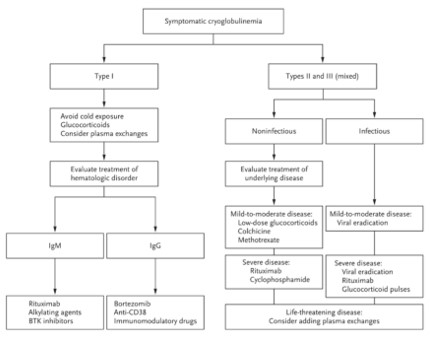
Figura 4
Algoritmo de tratamiento de las crioglobulinemias.
Los pacientes que presentan crioglobulinemia sintomática deben ser evaluados para determinar el tratamiento, prestando especial atención a las causas subyacentes. Para todos los pacientes con crioglobulinemia de tipo I, evitar la exposición al frío es una medida esencial, y las medias de compresión pueden ser suficientes para los pacientes que presentan solo púrpura. Los glucocorticoides y los intercambios plasmáticos suelen considerarse entre las estrategias de primera línea para los pacientes con una indicación farmacológica. La colaboración estrecha con los hematólogos es imperativa y, en ausencia de una indicación para el tratamiento de la afección hematológica subyacente, el tratamiento para la crioglobulinemia de tipo I debe determinarse en función del isotipo de la enfermedad.
El rituximab (un agente anti-CD20 de primera generación), los agentes alquilantes y los inhibidores de la tirosina quinasa de Bruton (BTK) son los preferidos para el tratamiento de la enfermedad mediada por IgM, mientras que el bortezomib (un inhibidor del proteasoma), los agentes anti-CD38 (p. ej., isatuximab y daratumumab) y los fármacos inmunomoduladores (p. ej., lenalidomida y talidomida) se han utilizado para la enfermedad mediada por IgG. Para las crioglobulinemias mixtas de tipo II y III, los regímenes terapéuticos se eligen en función de la gravedad de la vasculitis e implican la erradicación del desencadenante viral cuando la causa es infecciosa.
En la crioglobulinemia mixta relacionada con el virus de la hepatitis C, los agentes antivirales directos suelen ser suficientes para lograr una respuesta virológica sostenida y la remisión de la vasculitis en la enfermedad leve a moderada. Eventualmente, se podría prescribir concomitantemente un tratamiento con glucocorticoides a dosis bajas a corto plazo (es decir, ≤0,5 mg por kilogramo de peso corporal por día), aunque la mayoría de los pacientes se recuperan con un régimen sin glucocorticoides. Para las formas graves de la enfermedad, los agentes antivirales de acción directa deben combinarse con rituximab (375 mg por metro cuadrado de superficie corporal los días 1, 8, 15 y 22). En circunstancias potencialmente mortales (p. ej., glomerulonefritis con deterioro de la función renal, déficit motor, mononeuropatía múltiple, necrosis cutánea extensa o compromiso digestivo, cardíaco o pulmonar), los agentes antivirales de acción directa deben combinarse con glucocorticoides a dosis altas (es decir, 0,5–1,0 g de metilprednisolona durante 3 días consecutivos, seguido de dosis decrecientes de glucocorticoides orales) e intercambios plasmáticos.
En raras formas complicadas por un cáncer hematológico, se han utilizado con éxito combinaciones de rituximab, fludarabina y ciclofosfamida en centros especializados. En pacientes que no responden a los agentes antivirales de acción directa o que tienen una contraindicación para ellos, se puede utilizar rituximab solo para la inducción, seguido de una infusión de 500 mg cada 6 a 9 meses como tratamiento de mantenimiento. La crioglobulinemia mixta no infecciosa se puede controlar aún más con el uso del arsenal de tratamientos disponibles para la causa subyacente (p. ej., colchicina o metotrexato).