La esclerosis lateral amiotrófica (ELA) es una enfermedad mortal de la neurona motora , altamente heterogénea, con presentación clínica y contribuciones genéticas variables. Se han relacionado decenas de mutaciones genéticas con la ELA, lo que ha dado lugar a esfuerzos considerables para desarrollar un conjunto de herramientas de terapias dirigidas a genes. Sin embargo, es poco probable que estos tratamientos sean eficaces para la mayoría de los pacientes con ELA sin una causa genética conocida de la enfermedad. Si bien se sabe que numerosas vías moleculares están desreguladas en la ELA, lo que lleva a múltiples mecanismos propuestos que contribuyen a la muerte de las neuronas motoras, existe una decepcionante falta de opciones de tratamiento, incluso después de décadas de investigación. Por lo tanto, parece cada vez más probable que esta enfermedad heterogénea y compleja requiera enfoques de tratamiento multifactoriales, con terapias combinatorias desarrolladas sobre la base de la medicina personalizada. Por lo tanto, identificar y abordar vías moleculares relevantes en una amplia gama de pacientes con ELA, independientemente de la causa de la enfermedad, representa un área de mucho interés. Se han logrado avances activos en los numerosos aspectos desregulados de la patología de la ELA, desde el metabolismo energético hasta el procesamiento del RNA, la homeostasis de las proteínas y más.
Se sabe que las neuronas motoras en la ELA son vulnerables a la estimulación excesiva del glutamato, lo que llevó a la identificación temprana de la excitotoxicidad como factor impulsor de la enfermedad y el desarrollo de riluzol. Como una de las pocas opciones de tratamiento autorizadas para la ELA, el riluzol se dirige a la excitotoxicidad del glutamato, previniendo la muerte de las células de las neuronas motoras, aunque con mejoras limitadas en la esperanza de vida de los pacientes. La estimulación glutamatérgica está mediada en parte por los receptores de N-metil-D-aspartato (NMDAR) en las sinapsis (sNMDAR), pero el exceso de glutamato activa la señalización de muerte celular a través de NMDAR extrasinápticos (eNMDAR), que forman complejos con TRPM4. Dirigirse a los eNMDAR para prevenir la excitotoxicidad en las neuronas ha resultado un desafío hasta la fecha, en gran parte debido a la falta de especificidad del objetivo, ya que la interferencia farmacológica con la función sNMDAR puede ser perjudicial para la función sináptica normal. Trabajos anteriores del grupo Bading demostraron que un pequeño dominio intercelular dentro de TRPM4, denominado TwinF, es necesario para el complejo eNMDAR-TRPM4. Se derivaron computacionalmente pequeñas moléculas capaces de unirse específicamente a TwinF para alterar el complejo y así bloquear la señalización de muerte celular en presencia de glutamato. La especificidad de estas moléculas y su efecto neuroprotector al atacar eNMDAR-TRPM4 se demostró previamente mediante registro electrofisiológico, pérdida de coinmunoprecipitación del complejo y reducción de la muerte celular en modelos de lesión excitotóxica aguda. En este número de Cell Reports Medicine, Yan et al. describen FP802, un nuevo compuesto de interfaz TwinF potencialmente adecuado para uso clínico en ELA, con tentadores hallazgos preclínicos en modelos celulares y de ratón (Figura 1).
Yan et al. proporcionan evidencia de que FP802 mejora la patología en el modelo de ELA en ratones SOD1G93A. Para imitar mejor las condiciones clínicas, el tratamiento se inició al inicio de los síntomas motores. A pesar de este momento relativamente tardío de la intervención terapéutica, hubo un aumento significativo en la supervivencia, con fenotipos motores mejorados con respecto a los controles y preservación de las neuronas motoras en la médula espinal. Estos datos preclínicos demuestran que apuntar a los eNMDAR puede mejorar los resultados de enfermedades clave in vivo. Además, mejoraron los biomarcadores que se correlacionan con la progresión del fenotipo. Los marcadores neuroinflamatorios, aumentados en el ratón SOD1G93A debido a la muerte de la neurona motora, se redujeron en el grupo de tratamiento. También se redujo la cadena ligera de neurofilamentos séricos, un biomarcador clave para la ELA y un parámetro comúnmente medido en pacientes con ELA, allanando el camino para el seguimiento de la enfermedad en futuros ensayos clínicos. La farmacocinética mejorada de FP802 con respecto a compuestos anteriores, con un buen perfil de seguridad y falta de unión fuera del objetivo, también demuestra claramente que este fármaco es un fuerte candidato para avanzar hacia los ensayos clínicos. Sin embargo, aún quedan algunas preguntas. Aunque las formas de ELA dependientes de SOD1 son más vulnerables a la excitotoxicidad que otras formas de la enfermedad, se sabe desde hace mucho tiempo que el sistema de recaptación de glutamato, particularmente GLT-1, está desregulado en los pacientes con ELA. Por lo tanto, queda por establecer si el tratamiento con FP802 tiene el potencial de ser igualmente beneficioso en formas de ELA no SOD1. En este sentido, y también para probar la aplicabilidad del FP802 para su uso en humanos, Yan et al. generaron organoides del prosencéfalo a partir de células madre pluripotentes inducidas por humanos (iPSC). Los organoides derivados de pacientes con ELA, tomados de casos de ELA vinculados a SOD1 y esporádicos, fueron sobreestimulados con un exceso de NMDA, y se descubrió que ambos eran más susceptibles a la muerte celular que los controles sanos. Finalmente, FP802 fue neuroprotector contra esta agresión excitotóxica aguda, lo que demuestra una promesa inicial para el compuesto en un modelo humano in vitro junto con los datos in vivo en ratones.
La ELA, por su propia naturaleza, probablemente requerirá terapias combinatorias para afrontar los amplios desafíos que se presentan durante la patogénesis de la enfermedad. Yan et al. ofrecen un nuevo compuesto terapéutico, FP802, dirigido a un aspecto específico de la ELA, la excitotoxicidad, que tiene un potencial interesante en un enfoque de medicina combinatoria de este tipo. Por ejemplo, es posible imaginar un escenario en el que FP802 podría combinarse con otras terapias emergentes para proporcionar mejores resultados de tratamiento a los pacientes. Ejemplos de terapias emergentes que podrían ser apropiadas para uso combinatorio incluyen Tofersen, un oligonucleótido antisentido que reduce la producción de la proteína patológica SOD1 mutante en la SOD1-ELA familiar. Alternativamente, AMX0035 se dirige al estrés del retículo endoplásmico (RE) mitocondrial y retarda la progresión de los síntomas en pacientes con ELA tanto esporádica como familiar. De manera similar, el fármaco reutilizado terazosina aumenta la disponibilidad de energía para las neuronas motoras, proporcionando así neuroprotección. Dado el diverso mecanismo de acción de estas (y otras) terapias emergentes, un enfoque combinado podría proporcionar una opción de tratamiento sólida y modificadora de la enfermedad para un amplio espectro de pacientes con ELA. Por lo tanto, la identificación de FP802 representa un paso potencialmente importante hacia el futuro de terapias combinatorias efectivas para la ELA.
=> Recibir por Whatsapp las noticias destacadas
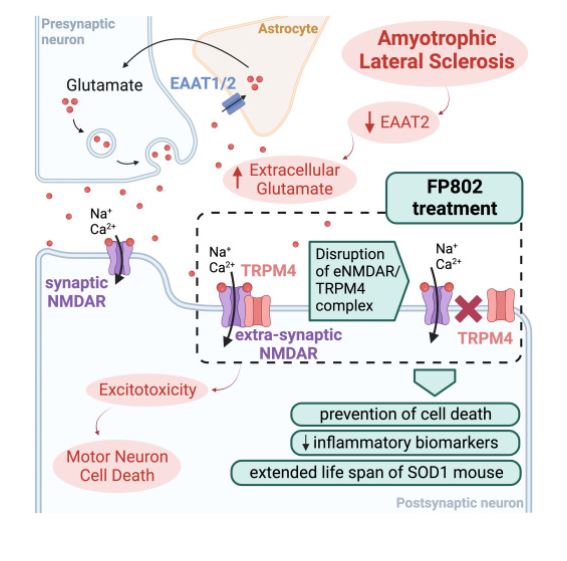
La figura 1FP802 altera específicamente el complejo entre los NMDAR extrasinápticos (eNMDAR) y TRPM4, mejorando los fenotipos del modelo de ELA al prevenir la excitotoxicidad.