Ronald Palacios Castrillo
Resumen
La edición del genoma ha sido una fuerza transformadora en las ciencias biológicas y la medicina humana, al ofrecer oportunidades sin precedentes para analizar procesos biológicos complejos y tratar las causas subyacentes de muchas enfermedades genéticas.
Las tecnologías basadas en CRISPR, con su notable eficiencia y fácil programación, están a la vanguardia de esta revolución. En esta revisión, Pacesa,et.al., (CELL.DOI:https://doi.org/10.1016/j.cell.2024.01.042 )analizamos el estado actual de las tecnologías de edición de genes CRISPR tanto en investigación como en terapia, destacando las limitaciones que las limitan y las innovaciones tecnológicas que se han desarrollado en los últimos años para abordarlas.
Además, examinaron y resumen el panorama actual de las aplicaciones de edición de genes en el contexto de la salud y la terapéutica humana. Finalmente, describen posibles desarrollos futuros que podrían dar forma a las tecnologías de edición de genes y sus aplicaciones en los próximos años.
=> Recibir por Whatsapp las noticias destacadas
Introducción
La edición del genoma (la modificación precisa y dirigida del material genético de los organismos vivos) representa uno de los avances más significativos en biología molecular. Tiene aplicaciones de gran alcance, desde desentrañar procesos biológicos fundamentales hasta impulsar avances en medicina, agricultura y biotecnología.
Con la aprobación de la primera terapia humana basada en CRISPR a finales de 2023, la edición del genoma CRISPR está entrando en una nueva era.
En esta revisión, Pacesa y colegas tienen como objetivo proporcionar una vista panorámica del panorama de la edición del genoma CRISPR, enfatizando su estado actual, los posibles desarrollos futuros y los obstáculos que deben superarse para hacer realidad su promesa para la medicina humana.
El pasado: desarrollo y limitaciones de la edición del genoma CRISPR
Antecedentes históricos
La edición del genoma surgió originalmente de avances en el campo de la reparación del DNA eucariótico. Estudios pioneros realizados a principios de la década de 1990 utilizando endonucleasas dirigidas como I-SceI, que reconoce secuencias de DNA de 18 pb, demostraron que la inducción de una rotura de doble hebra (DSB) dirigida en células de mamíferos estimulaba la recombinación homóloga en el sitio objetivo.
Esto estableció el concepto de edición del genoma utilizando nucleasas generadoras de DSB. La necesidad de diversas nucleasas con sitios de reconocimiento largos, que serían capaces de apuntar a sitios individuales en genomas eucariotas, estimuló el desarrollo posterior de enzimas nucleasas diseñadas basadas en fusiones complejas de endonucleasas de DNA no específicas con conjuntos en tándem de módulos de unión de DNA de secuencia específica.
Inicialmente se trataba de nucleasas con dedos de zinc (ZFN), introducidas a principios de la década de 2000, y más tarde de nucleasas efectoras similares a activadores de la transcripción (TALEN) en 2010-2011. Estos desarrollos sentaron las bases para la edición del genoma, pero el laborioso diseño y generación de Las ZFN y TALEN limitaron su uso.
El descubrimiento y desarrollo de nucleasas asociadas a CRISPR (Cas) guiadas por RNA marcó un cambio revolucionario debido a su simplicidad de programación, especificidad y versatilidad.
En 2007, se demostró que los sistemas procarióticos CRISPR-Cas funcionan como mecanismos de defensa del genoma adaptativo que reconocen y atacan ácidos nucleicos extraños asociados con virus (fagos) y otros elementos genéticos móviles.
En estos sistemas, los fragmentos de DNA invasor se adquieren y almacenan en matrices repetitivas, cuya transcripción y procesamiento posterior producen RNA CRISPR (crRNA).
Los crRNA funcionan como guías moleculares y programan maquinarias moleculares compuestas de proteínas Cas para reconocer los ácidos nucleicos invasores y atacarlos para su destrucción.
Investigaciones posteriores revelaron que los llamados sistemas CRISPR-Cas de tipo II escindían específicamente el DNA del fago, que la actividad dependía de la proteína Cas9 y que un componente adicional de RNA, el RNAcr transactivador (RNAtrancr), era esencial para la maduración del RNAcr.
Finalmente, estudios bioquímicos publicados en 2012 demostraron que Cas9 funcionaba como una endonucleasa que escinde el DNA, cuya especificidad estaba determinada por una estructura guía de RNA dual compuesta de RNAcr y TRAtracr.
El sistema CRISPR-Cas9 se simplificó aún más mediante la integración de tracrRNA y crRNA en un único RNA guía (sgRNA), proporcionando así un diseño de RNA de una nucleasa y una guía totalmente programable.
Esto se convirtió en la base para el desarrollo de tecnologías de edición del genoma CRISPR, mediante las cuales se podría lograr dirigir Cas9 a un sitio genómico específico mediante el diseño de un sgRNA con una secuencia coincidente.
El descubrimiento de la actividad de corte del DNA de Cas9 guiada por RNA desencadenó una carrera para reutilizarlo para la edición del genoma, que culminó con varios estudios publicados a principios de 2013 que informaron que la expresión de Cas9 y sgRNA específicos en células eucariotas condujo a la introducción de la modificación genética. en loci genómicos objetivo.
Edición del genoma basada en nucleasa CRISPR
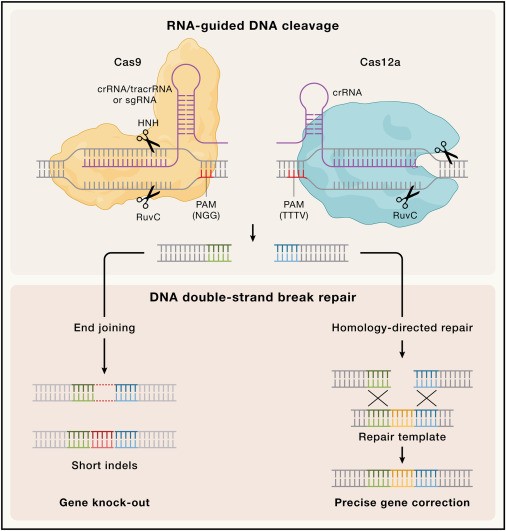
La programabilidad de las nucleasas CRISPR-Cas para generar roturas de DNA de doble cadena específicas de un sitio ha permitido su rápida adaptación a las tecnologías de edición del genoma (Figura 1).
La proteína Cas9 arquetípica que se origina en Streptococcus pyogenes (SpCas9), la primera nucleasa Cas reutilizada para la edición del genoma, sigue siendo el editor de genes más utilizado debido a su actividad y especificidad intrínsecamente altas.
Cas9 forma una nucleasa activa en asociación con cualquiera de los RNAcr. -complejos de RNAtracr o guías de RNAsg.
Para dirigir la nucleasa Cas9 al locus genómico de interés, se puede alterar la secuencia guía de 20 nt en el extremo 5′ del RNAcr para permitir el emparejamiento de bases canónicas con el objetivo de DNA.
La unión al objetivo depende además de la presencia de un motivo adyacente protoespaciador corto (PAM) ubicado en la cadena no objetivo (NTS) del DNA, inmediatamente aguas abajo del sitio objetivo.
El reconocimiento inicial del PAM da como resultado el desenrollamiento local del DNA objetivo, mientras que la base del RNA guía se empareja con la cadena objetivo (TS) del DNA en una dirección 5′-3′ comenzando en el extremo proximal del PAM del sitio objetivo, lo que desencadena cambios conformacionales en Cas9 que conducen a la activación del dominio de nucleasa.
Posteriormente, Cas9 escinde el sustrato de DNA bicatenario (dsDNA) tres nucleótidos aguas arriba de la secuencia PAM, generando DSB con extremos romos o salientes 5′ de un solo nucleótido.
La formación de DSB es catalizada por los dominios Cas9 HNH y RuvC, que escinden TS y NTS, respectivamente.
La inactivación selectiva de cualquiera de los dominios de nucleasa convierte Cas9 en nickasas guiadas por RNA, mientras que la inactivación de ambos dominios da como resultado una proteína de unión al DNA guiada por RNA que puede servir como plataforma para la entrega de proteínas fusionadas a loci genómicos específicos.
Cas12a, una nucleasa Cas procedente de sistemas CRISPR-Cas de tipo V, se descubrió unos años después de Cas9 y también se reutilizó para la edición del genoma.
A diferencia de Cas9, Cas12a no requiere un RNAtracr para su activación y, en cambio, cataliza el procesamiento nucleolítico de sus propias guías al reconocer una estructura de pseudonudo conservada en el segmento derivado de repetición del RNAcr, una característica que se ha aprovechado para la Edición multiplexada en vivo.
Cas12a se dirige a los DNA que contienen un TTTV PAM 5′-terminal y escinde ambas hebras dentro de la parte distal de PAM del sitio objetivo de manera secuencial utilizando su único sitio catalítico de dominio RuvC, lo que da como resultado la generación de 5-nt 5′ sobresale.
El producto PAM-DSB distal luego se disocia de la proteína, mientras que Cas12a permanece en un estado catalíticamente activo, capaz de escindir sustratos adicionales de DNA monocatenario ( ssDNA) en trans. Cas12a ha demostrado ser una nucleasa altamente eficiente capaz de edición genética precisa, con propiedades y funcionalidad complementarias a Cas9.
La actividad transnucleasa de Cas12a se ha utilizado además para la detección de ácidos nucleicos específicos de secuencia.
Los enfoques convencionales de edición del genoma se basan en la introducción de roturas de DNA de doble hebra específicas de un sitio en el genoma y su posterior resolución mediante vías endógenas de reparación del DNA celular (Figura 1).
Los DSB generados por las enzimas Cas9 o Cas12a generalmente se reparan mediante vías de unión de extremos, que suelen ser propensas a errores, o mediante mecanismos precisos de reparación dirigida por homología (HDR).
La unión de extremos es el modo predominante de reparación del DNA en células de mamíferos y se basa en la religación directa de los extremos del DNA roto mediante las vías de unión de extremos no homólogos (NHEJ) o unión de extremos mediada por microhomología (MMEJ).
El procesamiento de los extremos del DNA expuesto antes de que la religación conduzca a la adición o eliminación de nucleótidos, lo que resulta en inserciones o eliminaciones cortas (denominadas colectivamente indeles) en el sitio de la DSB, un resultado que se cree que se ve facilitado por la escisión repetida de las DSB reparadas con precisión hasta que los indeles acumulados impidan una mayor escisión.
Esto se usa más comúnmente para alterar selectivamente secuencias de genes que codifican proteínas para lograr inactivaciones o eliminaciones de genes mediante la introducción simultánea de dos DSB muy próximos. reproducible y depende del contexto de la secuencia local, que comprende inserciones de un solo nucleótido o pequeñas eliminaciones debidas a NHEJ, así como eliminaciones mediadas por MMEJ.
Por el contrario, HDR es una vía de reparación de DSB precisa que se basa en la presencia de una molécula de DNA homóloga para guiar el resultado de la reparación.
Al proporcionar de forma exógena una plantilla de reparación de homología artificial, se puede aprovechar HDR para introducir mutaciones, inserciones o eliminaciones deseadas precisamente dentro del locus genómico objetivo.
Las plantillas de reparación, entregadas como DNA bicatenario (normalmente a través de plásmidos o vectores virales) o como oligonucleótidos sintéticos de DNA monocatenario (ssDNA), llevan la mutación deseada flanqueada por secuencias homólogas a regiones a ambos lados de la DSB.
Aunque este enfoque en principio permite la edición con precisión de nucleótidos, HDR es principalmente activo solo en células que se dividen activamente, ya que requiere factores de reparación que comúnmente se expresan solo en las fases S y G2 del ciclo celular.
Por lo tanto, la eficiencia del resultado de HDR depende del tipo de plantilla de reparación, el método de administración, el tipo de célula, el contexto de cromatina local y otros factores que pueden afectar la elección de la vía de reparación del DNA para mejorar preferentemente el HDR y suprimir la reparación de la unión terminal.
Limitaciones de la edición del genoma CRISPR
La reutilización de los sistemas CRISPR-Cas como herramientas de edición de genes programables simples y eficaces ha hecho avanzar enormemente muchas áreas de la investigación básica y aplicada, sentando las bases para el desarrollo de terapias genéticas dirigidas y diversas aplicaciones biotecnológicas. Sin embargo, las características funcionales de un sistema de defensa biológica altamente evolucionado difiere de las funcionalidades esperadas de una herramienta precisa de edición del genoma.
En consecuencia, el potencial de aplicación de las herramientas de edición de genes basadas en CRISPR de primera generación está limitado por varios factores clave, siendo los principales la especificidad, el alcance de la focalización y la necesidad de depender de mecanismos endógenos de reparación de DSB para lograr ediciones genómicas (Figura 2). Finalmente, la entrega de componentes CRISPR está limitada por limitaciones específicas de los vectores de entrega y las células u organismos diana.
Los sistemas CRISPR-Cas naturales toleran, hasta cierto punto, desajustes entre el RNA guía y el objetivo, una probable consecuencia evolutiva de la necesidad de contrarrestar la alta tasa de mutación de los fagos. Sin embargo, esta propiedad no es deseada para aplicaciones de ingeniería genómica, ya que puede dar lugar a la selección y edición de sitios fuera del objetivo parcialmente complementarios en otras partes del genoma, además del locus objetivo previsto.
La actividad fuera del objetivo de Cas9 ha sido documentada por numerosos estudios que muestran que la enzima tolera un número considerable y una variedad de desajustes de nucleótidos dentro del heterodúplex guía-objetivo de una manera dependiente de la guía. Los objetivos pueden variar desde sitios que albergan una única base errónea hasta objetivos que contienen múltiples desajustes consecutivos, o incluso inserciones o eliminaciones de nucleótidos. no da como resultado la escisión y edición del dsDNA debido a puntos de control intrínsecos en el mecanismo de unión y escisión del DNA de Cas9.
Además, los estudios de perfiles fuera del objetivo han demostrado que la frecuencia de eventos de escisión fuera del objetivo es consistentemente menor in vivo en comparación con el DNA genómico aislado, lo que sugiere que factores adicionales, incluida la estructura del genoma, podrían gobernar la actividad de edición de Cas9 en las células.
Sin embargo, la escisión simultánea fuera del objetivo en múltiples sitios dentro del genoma puede, en última instancia, dar como resultado reordenamientos genómicos como deleciones.,inversiones o translocaciones cromosómicas y desencadenan daños en el DNA y vías de respuesta al estrés.
La edición fuera del objetivo sigue siendo una preocupación importante para las aplicaciones terapéuticas y ha impulsado grandes esfuerzos para desarrollar métodos robustos y sensibles para la predicción y detección de ediciones fuera del objetivo y para mejorar la especificidad de los editores del genoma CRISPR mediante ingeniería molecular.
Ámbito de orientación: requisitos de PAM
El mecanismo de unión al DNA de las nucleasas CRISPR restringe su alcance de orientación a sitios genómicos diana flanqueados por una secuencia PAM.
Aunque la NGG PAM de SpCas9, la nucleasa editora del genoma más utilizada, teóricamente permite encontrar un sitio objetivo adecuado cada 8 nucleótidos en promedio, SpCas9 no puede seleccionar fácilmente algunas regiones genómicas debido a un alto contenido de A/T.
Se han identificado y adoptado varios ortólogos de Cas9 naturales con especificidades PAM alternativas para la edición del genoma; sin embargo, muchos de ellos tienen requisitos de PAM aún más restrictivos.
Aunque esto proporciona una mayor especificidad de orientación y reduce la actividad fuera del objetivo, a menudo da como resultado eficiencias de edición y escisión del DNA subóptimas.
Las nucleasas Cas12a reconocen secuencias PAM ricas en T, típicamente TTTV, pero su alcance de orientación también está restringido. Para superar la limitación de los requisitos de PAM, en los últimos años se han desarrollado una serie de variantes artificiales de Cas9 con especificidades de PAM modificadas o relajadas, como se describe en la siguiente sección.
Aunque estos amplían en gran medida la gama de sitios a los que se puede dirigir, la orientación PAM relajada se asocia con una fuerte disminución en la especificidad de la orientación, lo que aumenta la probabilidad de efectos fuera del objetivo y una reducción de la eficiencia de la edición en el objetivo debido al secuestro en sitios fuera del objetivo.
Controlar los resultados de la edición
La generación de DSB dentro del genoma utilizando nucleasas específicas mejora significativamente la tasa de HDR en células de mamíferos.
A pesar de esto, el uso de HDR está restringido a células en división y a menudo produce resultados de edición heteroalélica debido a la edición simultánea resultante de las vías de unión de extremos.
Además, la precisión de la edición en el sitio objetivo genómico previsto está limitada por otros resultados adversos, incluidos grandes deleciones y reordenamientos cromosómicos e incluso pérdida de cromosomas.
Como resultado, mucha investigación se ha dedicado a desarrollar enfoques para controlar los resultados de la reparación del DNA, particularmente para estimular la tasa de reparación de HDR y así aumentar la eficiencia de las mutaciones knocking (inserción).
Lo más importante es que se ha demostrado que la elección de la plantilla de reparación y su administración afecta en gran medida la eficiencia de HDR.
Los métodos para mejorar HDR incluyen el uso de plantillas ssDNA asimétricas, la introducción de mutaciones silenciosas para bloquear la escisión recurrente en el sitio objetivo, o la fijación de la plantilla del donante de reparación al sitio de rotura.
Además, la manipulación del ciclo celular combinada con la administración de ribonucleopartículas Cas9 preensambladas también da como resultado una HDR mejorada.
La sincronización del ciclo celular se puede lograr mediante el uso de inhibidores de moléculas pequeñas para ralentizar temporalmente la fase S o aumentando la proporción de células en la fase G2/M. Otros métodos se centran en cambiar el equilibrio entre la elección de la vía NHEJ y HDR hacia HDR mediante la supresión de factores clave de NHEJ.
De manera análoga, la asociación directa de Cas9 con factores de reparación involucrados en HDR, como Rad51, Rad52 o Mre11, puede mejorar sustancialmente las tasas de mutaciones knocking.
En células T humanas primarias, se podrían lograr eficiencias de edición HDR superiores al 80% -90% con plantillas de reparación de ssDNA diseñadas para formar extremos de dsDNA reconocibles por Cas9-RNP, utilizadas junto con moduladores de reparación de ADN de molécula pequeña.
Finalmente, estudios recientes han demostrado que la eficiencia de HDR se puede mejorar reorientando los subproductos de edición de NHEJ utilizando RNA guía secundarios.
A pesar de estos avances, el proceso de introducción de mutaciones knocking, particularmente inserciones largas, utilizando plantillas de homología sigue siendo un desafío. Las células posmitóticas, terminalmente diferenciadas, como las neuronas, en las que el HDR no se produce en niveles apreciables, siguen siendo en gran medida recalcitrantes a la edición precisa utilizando estrategias canónicas basadas en DSB.
Estas limitaciones, junto con los posibles efectos genotóxicos de los DSB, han motivado el desarrollo de tecnologías de edición del genoma que no dependen de DSB y obvian la necesidad de HDR, incluida la edición base y la edición principal y, más recientemente, recombinasas y transposasas basadas en CRISPR.
Entrega
La entrega dirigida de editores de genes sigue siendo el factor limitante para la mayoría de las aplicaciones de edición de genes in vivo y ex vivo.
En particular, la entrega segura, específica y eficiente de componentes CRISPR a las células específicas es un requisito previo para una edición terapéutica exitosa del genoma. Además, la inmunogenicidad de los componentes CRISPR y sus vectores de administración presenta una preocupación para las aplicaciones terapéuticas in vivo.
Se han identificado anticuerpos anti-Cas9 preexistentes y células T reactivas en humanos, y la inmunidad Cas9 se ha asociado con resultados terapéuticos comprometidos en modelos de enfermedades de primates caninos y no humanos.
Se han propuesto varias estrategias para superar la inmunidad preexistente. , por ejemplo, diseñar Cas9 para eliminar epítopos inmunogénicos, modular las respuestas inmunes y limitar la duración de la expresión de Cas9.
Las enzimas Cas9/Cas12a y sus RNA guía se pueden administrar en múltiples formatos, según el tipo de célula u organismo objetivo.
Para la mayoría de las aplicaciones in vitro (es decir, ex vivo) en células cultivadas, la electroporación (nucleofección) o la transfección mediada por liposomas siguen siendo la modalidad de administración más utilizada debido a su alta eficiencia.
Cas9 y los componentes de RNA guía se pueden administrar como RNA, DNA plasmídico o complejos de ribonucleoproteína (RNP) reconstituidos in vitro.
La administración transitoria basada en RNP se ha convertido en la opción preferida de edición de genes para aplicaciones terapéuticas ex vivo, ya que la expresión a largo plazo del complejo Cas9 a partir de un plásmido puede dar como resultado altas tasas de edición fuera del objetivo e integración aleatoria de plásmidos.
Para la edición del genoma de la línea germinal en muchos organismos modelo, las RNP Cas9 o el mRNA generalmente se administran mediante microinyección o electroporación.
La administración in vivo de CRISPR-Cas9 en células de mamíferos se logra comúnmente utilizando vectores virales.
Se pueden diseñar adenovirus, lentivirus y virus adenoasociados (AAV) para reemplazar los genes virales dentro del vector con módulos de edición de genes.
Los AAV siguen siendo los vectores preferidos para la administración in vivo debido a su baja inmunogenicidad, alta eficiencia de transducción y diverso tropismo celular.
Sin embargo, los AAV son virus relativamente pequeños con una capacidad de embalaje de carga limitada (aproximadamente 4,7 kb).
Como resultado, es difícil empaquetar los genes que codifican SpCas9 (4,2 kb) y su sgRNA (~100 nt) en un único vector AAV a menos que se utilicen promotores ultracompactos.
Para superar esta limitación, se ha centrado mucho en adaptar los genes más pequeños. Ortólogos de Cas9, y enzimas compactas de la familia Cas12.
Por último, el mRNA de Cas9 en combinación con RNA guía sintéticos, o partículas RNP reconstituidas in vitro, se pueden administrar a células in vivo utilizando enfoques no virales como las nanopartículas basadas en lípidos (LNP).
La ventaja de estos métodos es que son generalmente son más seguros y exhiben una menor inmunogenicidad que los vectores basados en virus.
Los LNP se internalizan mediante endocitosis, tras la cual el contenido de LNP escapa del endosoma y se transfiere al núcleo, o es degradado por el lisosoma, lo que limita su eficiencia. Recientemente han surgido nuevas formulaciones de LNP que dan como resultado una mayor eficiencia en la focalización de órganos y tejidos específicos.
En conjunto, la administración de Cas9 y otros editores de genoma utilizando nanopartículas lipídicas se ha aplicado con éxito en una variedad de tipos de células y organismos.
El presente: Tecnologías actuales y sus aplicaciones
Desde la primera demostración de edición de genes basada en CRISPR, el campo ha evolucionado a un ritmo sin precedentes.
Las capacidades de los editores de genoma dependientes de DSB de primera generación basados en nucleasas Cas9 y Cas12a se han mejorado mediante innovaciones continuas que no solo han aumentado la versatilidad de estas herramientas sino que también han refinado su precisión y minimizado las consecuencias de edición no deseadas.
Sin embargo, persisten las preocupaciones sobre su seguridad, tanto debido a la actividad de edición fuera del objetivo como a los posibles efectos genotóxicos de los DSB en el objetivo, incluida la inducción de p53.
Para reducir la aparición de ediciones no deseadas, se han explorado varios enfoques para el control espaciotemporal preciso de los editores del genoma CRISPR, por ejemplo, el uso de inhibidores de proteínas anti-CRISPR naturales para la edición restringida a tejidos.
Los DSB y la necesidad de abordar la baja eficiencia de HDR han impulsado además el desarrollo de tecnologías CRISPR de “segunda generación” que median en la edición del genoma sin depender de la formación de DBS y HDR, en particular editores base (BE) y editores principales (PE).
El panorama actual y ampliado de tecnologías disponibles (Figura 3) ofrece, por lo tanto, un enfoque mucho más personalizado para la edición del genoma, con tecnologías específicas particularmente adecuadas para ciertos tipos de ediciones o modalidades de entrega.
Aunque estas adiciones al conjunto de herramientas de edición del genoma han contribuido significativamente a abordar las muchas limitaciones asociadas con la edición del genoma CRISPR canónico, todavía tienen límites en sus actividades, especificidad y entrega.
Esta sección describe cómo han surgido las tecnologías actuales, qué limitaciones han ayudado a mitigar y qué limitaciones aún enfrentan.
Nucleasas más allá de Cas9
El desarrollo inicial de Cas9 como nucleasa editora del genoma motivó estudios de seguimiento destinados a identificar nuevas enzimas Cas de origen natural con posibles aplicaciones en la ingeniería genómica. Gracias a estos esfuerzos, en los últimos años se han descubierto y adoptado para la edición del genoma más de una docena de nuevas nucleasas guiadas por RNA evolutivamente diversas.
El descubrimiento de Cas12a en 2015 marcó una expansión fundamental más allá de Cas9, ofreciendo una nucleasa alternativa con un requisito de PAM distinto, un formato de RNA guía y un patrón de escisión de DNA. Se ha informado que las enzimas Cas12a exhiben una mayor especificidad y una menor actividad fuera del objetivo in vivo, en parte debido a sus velocidades más lentas de escisión de DNA.
Exploraciones bioinformáticas posteriores han descubierto una amplia gama de efectores Cas de tipo V con distintos requisitos de PAM y RNA guía.
Se necesitan vectores virales de tamaño limitado (como AAV). Finalmente, las nucleasas guiadas por RNA dirigidas a RNA, como Cas13, Cas12a2 y Cas12g, han introducido una nueva modalidad en la caja de herramientas de edición molecular, lo que permite la degradación del objetivo o edición de mRNA, y detección de ácidos nucleicos.
Aunque estos descubrimientos han generado nuevas capacidades, cada nucleasa Cas viene con su propio conjunto de limitaciones, incluida la orientación restringida a PAM, actividades variables in vivo y perfiles fuera del objetivo, o potencial inmunogenicidad.
Sin embargo, la disponibilidad de múltiples enzimas ahora permite adoptar un enfoque más personalizado para la edición del genoma, lo que permite flexibilidad para diversas aplicaciones. La ortogonalidad de muchas de estas enzimas con respecto a sus RNA guía es particularmente útil para aplicaciones de multiplexación.
Variantes de Cas9 de alta fidelidad
Para abordar el problema de la actividad fuera del objetivo, se han desarrollado variantes diseñadas de SpCas9 con especificidad mejorada utilizando dos enfoques complementarios.
El primero implica el diseño racional basado en la estructura de mutaciones que mejoran la fidelidad, basado en la idea de que la eliminación de contactos específicos entre la proteína Cas9 y el DNA objetivo unido hace que el complejo Cas9-RNA guía sea más sensible a los desajustes en el DNA sustrato y, por lo tanto, reduce la probabilidad. de unión y escisión fuera del objetivo.
Los estudios biofísicos y bioquímicos de estas variantes han revelado que las mutaciones ralentizan sustancialmente la tasa de escisión del DNA, promoviendo así la liberación fuera del objetivo. El segundo enfoque utiliza métodos de evolución dirigida para seleccionar mutaciones que reducir la edición fuera del objetivo.
Se han realizado esfuerzos similares para diseñar variantes de alta fidelidad de otras enzimas Cas9 y Cas12a. Aunque las variantes de alta fidelidad desarrolladas hasta la fecha ofrecen especificidades considerablemente mayores que las enzimas de tipo salvaje, sus eficiencias pueden variar entre diferentes objetivos y aplicaciones de DNA.
Además, como cada objetivo está asociado con un conjunto único de objetivos fuera de objetivo con frecuencias de edición variables. «Es probable que ninguna de las variantes de nucleasa de alta fidelidad disponibles actualmente sea de aplicación universal».
Guía de modificaciones del RNA.
La ingeniería de los RNA guía de los editores del genoma para aumentar la especificidad ofrece una alternativa convincente al empleo de mutantes de nucleasa de alta fidelidad. El primero de estos esfuerzos se centró en el uso de RNA guía truncados para Cas9, en los que el segmento guía se trunca de 20 a 17-18 nucleótidos.
Se ha demostrado que estos reducen significativamente la actividad fuera del objetivo de SpCas9 en una variedad de sitios, pero también muestran una menor eficiencia o activan la edición en nuevos sitios fuera del objetivo.
Modificaciones del extremo 5′ del segmento guía, ya sea mediante la introducción se ha demostrado que las estructuras secundarias o los nucleótidos desapareados mitigan el reconocimiento fuera del objetivo.
Varios estudios han demostrado la funcionalidad de las guías de RNA-DNA «híbridas» y han descubierto que las sustituciones de 2′-desoxinucleótidos dentro del segmento guía aumentan sustancialmente la especificidad de la edición de genes en las células.
La introducción de otras modificaciones químicas como 2′- Los nucleótidos O-metilo o 2′-fluoro y los enlaces fosforotioato dentro del RNA guía también han surgido como una estrategia poderosa para aumentar la especificidad de Cas9 y mejorar la estabilidad de la guía.
Algunas de estas modificaciones han mostrado una sólida actividad de edición de genes in vivo, pero los efectos de los nucleótidos modificados dependen en gran medida de la posición, de forma similar a las sustituciones del DNA.
Editores alternativos del genoma PAM
Para superar las limitaciones del sitio objetivo dependiente de PAM de las nucleasas Cas, varios estudios han buscado ampliar el alcance de direccionamiento de PAM de SpCas9, ya sea mediante ingeniería racional basada en la estructura o evolución dirigida para introducir sustituciones de aminoácidos que alteren la especificidad en el dominio que interactúa con PAM.
Inicialmente, esto condujo al desarrollo de variantes VQR, EQR y VRER SpCas9 capaces de apuntar a PAM NGAN, NGNG y NGCG, respectivamente. La selectividad PAM de SpCas9 se ha relajado aún más en variantes de ingeniería adicionales dirigidas a PAM no G.
Para hacer que CRISPR-Cas9 pueda acceder a una gama aún más amplia de sitios genómicos, esfuerzos recientes lograron diseñar variantes de SpCas9 capaces de apuntar a NRN PAM, incluidos diseños de Cas9 casi «sin PAM».
Las variantes son muy variables, han ampliado significativamente el potencial de direccionamiento de Cas9 y han simplificado la selección del sitio objetivo, complementando SpCas9 de tipo salvaje y otros ortólogos de Cas9 de origen natural. Del mismo modo, también se han desarrollado variantes de Cas12a con especificidades PAM relajadas para aplicaciones de edición.
Edición básica
Los editores de base (BE) derivados de CRISPR se han desarrollado como una tecnología versátil para generar mutaciones puntuales específicas sin la necesidad de generar DSB y proporcionar plantillas de reparación de homología, lo que permite la edición en células con deficiencia de HDR.
Los BE son fusiones modulares de un RuvC- Versión nickasa inactivada de Cas9 con una enzima nucleótido desaminasa. Inicialmente, se desarrollaron dos clases de BE.
Figura 1 Principios moleculares de la edición del genoma CRISPR.
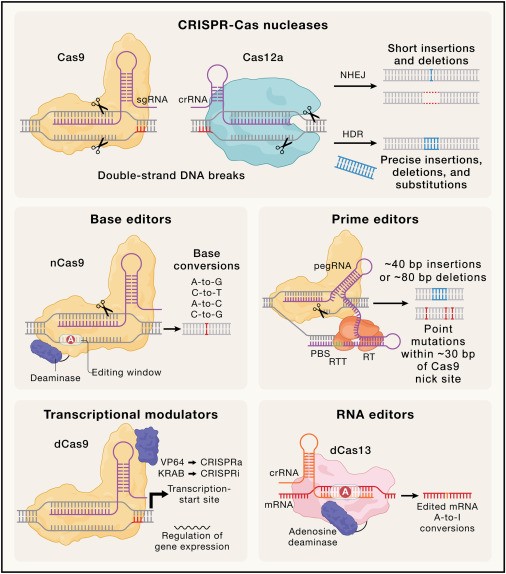
Los BE de citosina (CBE), que contienen dominios catalíticos derivados de citidina desaminasas como APOBEC1, así como un dominio inhibidor de la uracilo glicosilasa (UGI), median la conversión de C a T. A su vez, los BE de adenina (ABE) generan conversiones de A a G utilizando un dominio de adenosina desaminasa de la desaminasa específica de RNATadA que ha sido diseñada mediante evolución dirigida para actuar sobre el ssDNA.
Tras la unión del módulo Cas9, los BE desaminan una citosina o adenina dentro de una «ventana de edición» en el segmento distal PAM de la cadena de DNA no objetivo desplazada a uracilo o inosina, respectivamente. Estos se leen durante la replicación del DNA como timina y guanina, respectivamente, induciendo mutaciones en puntos de transición.
Desde su invención, los editores CBE y ABE originales han pasado por varias iteraciones de diseño para mejorar la actividad y reducir la cantidad de ediciones fuera de objetivo inducidas por la desaminasa, y también se han desarrollado Cas12a BE.
El repertorio de edición básico también se ha ampliado para cubrir también las transversiones de A a C, de A a Y y de C a G. Debido a sus resultados de edición en gran medida predecibles, los BE se han aplicado para la detección de mutaciones y la desactivación de todo el genoma.
La precisión de los BE los hace adecuados para correcciones terapéuticas de enfermedades causadas por mutaciones de un solo punto.
Aunque los BE permiten un control más preciso sobre los resultados de la edición, adolecen de varias limitaciones.
Estos incluyen eficiencia limitada, edición por espectadores, ventanas de edición amplias y actividad sustancial fuera del objetivo. Las variantes ABE de última generación exhiben mayores eficiencias de edición y frecuencias más bajas de edición fuera del objetivo independiente de Cas9 que los CBE, lo que ha provocado la ingeniería de nuevas variantes de CBE mediante la evolución dirigida de ABE.
Figura 2 Limitaciones de la edición del genoma CRISPR
Actividad fuera del objetivo
Finalmente, tanto los ABE como los CBE pueden tener efectos genotóxicos no deseados al generar DSB, deleciones y translocaciones en el locus objetivo, aunque a frecuencias más bajas que la Cas9 canónica basada en nucleasa edición. Estos pueden mitigarse parcialmente modulando el tiempo de entrega y los niveles de expresión del editor.
Edición principal
La edición Prime es un enfoque basado en Cas9 desarrollado para generar mutaciones puntuales específicas, así como inserciones o eliminaciones de manera independiente de HDR.
El editor principal consta de un RNA guía de edición principal (pegRNA) y una construcción de proteína de fusión compuesta de nickasa Cas9 con un dominio HNH inactivado y un dominio de transcriptasa inversa (RT) diseñado.
El pegRNA contiene una extensión de secuencia terminal 3′ que es complementaria a la NTS de la diana genómica deseada y contiene las mutaciones deseadas. Cas9 genera una mella en el NTS, que luego se empareja con la extensión complementaria de pegRNA.
Luego, la mutación se introduce mediante la extensión catalizada por RT del extremo 3′ del NTS utilizando el pegRNA como plantilla. A esto le sigue el nuevo recocido de las cadenas de DNA para producir un intermedio de solapa 5′ que se somete a escisión y ligadura, fijando la edición en el DNA genómico.
Esta síntesis de cadena dirigida permite la introducción de inserciones de hasta 40 pb o eliminaciones de hasta 80 pb de longitud, así como mutaciones puntuales de hasta 30 pb desde el sitio de mellado de Cas9.
A partir del diseño del editor principal de primera generación, en el que se fusionó una nickase Cas9 con una RT de tipo salvaje del virus de la leucemia murina Moloney (MMLV), las generaciones posteriores de diseños de PE trajeron mejoras en la eficiencia de la edición principal al incluir dominios RT MMLV diseñados con termoestabilidad mejorada e introducción de un segundo sgRNA que genera una mella en la cadena no editada para promover la retención de la edición en el DNA genómico.
Se han logrado mejoras adicionales inhibiendo la vía de reparación de desajustes del DNA, así como mejorando la localización nuclear, la expresión y el corte del DNA. Además, se ha añadido estructuras secundarias estabilizadoras al extremo 3′ del pegRNA, que contrarresta su degradación.
Se ha demostrado que mejora la eficiencia de la edición principal. Hasta la fecha, la edición primaria se ha aplicado con éxito en una variedad de organismos y tipos de células. Sin embargo, dependiendo de la edición prevista, la secuencia del sitio objetivo y el tipo de célula, las eficiencias de la edición primaria son muy variables y, a menudo, bajas.
Además, la edición principal no evita por completo la generación de DSB en el objetivo, lo que produce efectos no deseados y potencialmente genotóxicos.
Para abordar estas limitaciones, se continúan desarrollando nuevas variantes de PE con rendimiento mejorado, incluidos sistemas que permiten la generación de ediciones más extensas utilizando pares de pegRNA.
Modulación transcripcional: CRISPRi y CRISPRa
Las tecnologías CRISPR no sólo son aplicables para la edición del genoma sino que también han permitido la manipulación transitoria de la expresión genética.
Inicialmente se utilizaron mutantes catalíticamente inactivos de Cas9 en bacterias para atacar promotores de genes y bloquear estéricamente las RNA polimerasas, inhibiendo así la transcripción de RNA.
Para suprimir la expresión génica en células eucariotas, Cas9 inactivo para nucleasas puede fusionarse con varios moduladores transcripcionales y epigenéticos, por ejemplo, el dominio represor transcripcional KRAB, y dirigirse a regiones promotoras de genes transcritos activamente.
El enfoque resultante, conocido como interferencia CRISPR (CRISPRi), permite eliminar eficientemente la expresión genética como una alternativa a la interferencia de RNA pequeña basada en RNA de interferencia (RNAnip).
De manera análoga, las fusiones de proteínas Cas9 sin nucleasas se pueden usar para activar la expresión de genes específicos, ya sea reclutando directamente factores de activación transcripcional o modulando el estado de la cromatina.
La activación CRISPR (CRISPRa) se ha logrado fusionando dCas9 con dominios de transactivación como VP64 y sus derivados.
Alternativamente, las fusiones con enzimas modificadoras de histonas, como desmetilasas o acetilasas, pueden alterar de manera específica los marcadores epigenéticos en un sitio particular. e inducir estados activos de cromatina para impulsar la expresión génica.
Dichos enfoques se han adaptado fácilmente para la detección de pérdida de función de todo el genoma (CRISPRi) o ganancia de función (CRISPRa).
Sin embargo, como la unión del DNA por Cas9 es generalmente más promiscua que la escisión, la modulación transcripcional dirigida por CRISPR adolece de una considerable actividad fuera del objetivo.
Silenciamiento y modificaciones de RNA dirigidos.
El descubrimiento de enzimas Cas13 dirigidas a ssRNA guiadas por RNA de sistemas CRISPR-Cas tipo VI ha llevado al desarrollo de herramientas moleculares para el silenciamiento dirigido de RNA, así como para la edición y modificación. Como las ediciones de RNA son transitorias, la edición del transcriptoma ofrece potencialmente una alternativa atractiva a la edición del genoma en determinados contextos.
Por ejemplo, en enfermedades agudas como dolor, inflamación o infecciones virales, la edición transitoria de RNA podría proporcionar efectos terapéuticos temporales sin introducir cambios genéticos permanentes.
Los enfoques actuales de edición de RNA se basan en la fusión de proteínas dCas13 inactivas con nucleasas con adenosina desaminasa que actúa sobre enzimas de RNA (ADAR) para catalizar la desaminación de adenosina dentro de las moléculas de RNA diana, generando una base de inosina. La hibridación del RNA guía Cas13 con el RNA objetivo produce un sustrato de RNAbc para la enzima ADAR.
La inosina resultante puede emparejarse efectivamente con una citosina y, por lo tanto, se lee como guanosina durante la traducción, lo que resulta en la introducción de cambios de codones en el mRNA equivalente a una conversión de A a G.
Los esfuerzos posteriores de ingeniería de proteínas han dado como resultado el desarrollo de una variante de ADAR2 capaz de realizar la conversión de C a U en RNA guiada por Cas13. El concepto de modificación dirigida del RNA mediante fusiones de proteínas dCas13 se ha ampliado aún más para desarrollar «editores epitranscriptómicos» capaces de instalar de forma específica la modificación de N6-metiladenosina (m6A) en los mRNA objetivo.
Aplicaciones actuales en investigación básica y medicina humana.
Los avances tecnológicos en la edición del genoma CRISPR han dado lugar a una serie de aplicaciones con un potencial significativo para mejorar la salud humana, que van desde avances en la investigación fundamental hasta el desarrollo de nuevos tratamientos terapéuticos (Figura 4).
En primer lugar, CRISPR ha transformado la investigación genética al permitir a los científicos imitar mutaciones que causan enfermedades en varios modelos experimentales, crear métodos de detección a gran escala de todo el genoma y desarrollar dispositivos de registro genético sintético para estudiar el desarrollo normal y la progresión de la enfermedad.
Aunque no se analizan aquí en detalle, los sistemas CRISPR se han reutilizado para desarrollar diagnósticos moleculares que permitan una detección específica, rápida y sensible de DNA o RNA viral. Las tecnologías CRISPR también se han utilizado para establecer estrategias para la eliminación de patógenos humanos virales o bacterianos, estos últimos mediante el desarrollo de bacteriófagos diseñados.
Un ejemplo específico de limitación de la propagación de patógenos son los impulsores genéticos basados en CRISPR, en los que rasgos supresores particulares, como como la esterilidad femenina, se introducen para colapsar las poblaciones de insectos que albergan patógenos, principalmente los mosquitos transmisores de la malaria.
Finalmente, la última década de edición del genoma CRISPR ha culminado con el desarrollo de una multitud de enfoques terapéuticos para enfermedades genéticas, varios de los cuales han pasado de estudios preclínicos en modelos celulares y animales a ensayos clínicos en humanos. Esto incluye estrategias de corrección terapéutica tanto in vivo como ex vivo.
Los enfoques de corrección terapéutica in vivo implican la administración de componentes de edición de genes a los tejidos afectados dentro del cuerpo humano. Por el contrario, los enfoques ex vivo implican recolectar células de un paciente, editarlas en un laboratorio y luego trasplantar las células editadas al paciente.
Además, la edición CRISPR ex vivo ha permitido la generación de terapias celulares autólogas y alogénicas modificadas con el genoma, destinadas principalmente a la inmunoterapia contra el cáncer.
Genética directa: pantallas CRISPR, rastreo de linaje y registradores moleculares
Los enfoques genéticos avanzados, como las pruebas de eliminación de todo el genoma, son herramientas poderosas para investigar las funciones biológicas en la salud y la enfermedad. Aprovechando los avances en curso en la edición del genoma, las pantallas CRISPR permiten la perturbación sistemática de miles de genes individuales y elementos genómicos no codificantes dentro de las células, facilitando la identificación de genes asociados con vías biológicas específicas y sus interacciones.
En estas pantallas de alto rendimiento, las bibliotecas de ARN guía junto con las enzimas Cas se administran a las células, generalmente mediante transducción con un conjunto de vectores lentivirales, lo que garantiza que cada célula reciba un sgRNA distinto.
Al aplicar la selección basada en la supervivencia celular o en la lectura, se utiliza la secuenciación de próxima generación para identificar qué ARN guía se enriquecen o, por el contrario, se eliminan de la población celular, identificando así los genes diana vinculados al fenotipo específico.
Este enfoque general se ha adaptado para utilizar diversos tipos de tecnologías CRISPR, incluidas las nucleasas CRISPR para pantallas knockout, CRISPRi, CRISPRa, BE, PE, así como Cas13, lo que permite estudiar una amplia gama de modificaciones genéticas. Las primeras pantallas CRISPR agrupadas se diseñaron para identificar genes que contribuyen a fenotipos fácilmente seleccionables, como el crecimiento celular o la resistencia a los medicamentos.
Las pantallas CRISPR multiplexadas también se han utilizado para descubrir interacciones genéticas por pares que suprimen el crecimiento de células cancerosas. Además, se combinaron in vivo. También se han realizado pruebas CRISPR en embriones de pez cebra, inactivando genes individuales y aislando embriones con fenotipos específicos para identificar las alteraciones genéticas responsables.
Los BE, que permiten la introducción eficiente de mutaciones puntuales, se han convertido en herramientas particularmente poderosas para la detección de variantes genéticas tanto con pérdida como con ganancia de función.
La integración de pantallas CRISPR con metodologías ómicas unicelulares ha mejorado enormemente nuestra capacidad para estudiar la función genética. Métodos como PERTURB-seq, CRISP-seq, CROP-seq y Mosaic-seq utilizan la secuenciación de ARN unicelular para rastrear los sgRNA en células individuales y monitorear simultáneamente el espectro completo de cambios en la expresión genética después de las perturbaciones inducidas por CRISPR.
Mayores refinamientos en estas metodologías han permitido la investigación directa de la función genética in vivo, como el análisis de genes asociados con trastornos del desarrollo neurológico en el cerebro del ratón. Además, estos métodos se han combinado con secuenciación de ARN unicelular resuelta espacialmente para analizar las intrincadas interacciones entre el cáncer y las células inmunes en los tumores, o se han vinculado con espectrometría de masas unicelular para obtener perfiles detallados de expresión de proteínas.
Más allá de las pantallas de todo el genoma, las herramientas de edición del genoma CRISPR también han facilitado el desarrollo de enfoques de rastreo de linajes para permitir el seguimiento de la proliferación celular y la reconstrucción de árboles de linajes celulares.
El rastreo del linaje se logra mediante la introducción de códigos de barras de ADN artificiales en las células y su edición continua mediante Cas9 a lo largo del tiempo, que se detecta mediante secuenciación de ARN unicelular para mapear filogenias celulares e integrarlas con resultados transcriptómicos unicelulares.
Por ejemplo, estos enfoques han permitido la reconstitución exitosa de linajes celulares durante el desarrollo del pez cebra y la embriogénesis del ratón, y han rastreado la evolución y la dinámica de las células cancerosas metastásicas.
La edición del genoma CRISPR ha permitido el desarrollo de dispositivos de memoria sintética para registrar información de eventos moleculares transitorios en códigos de barras de ADN.
Esto incluye información sobre la historia transcripcional pasada, la actividad de elementos reguladores cis específicos o la exposición a ciertos factores ambientales. Dichos registradores moleculares funcionan vinculando un evento molecular particular con la generación de una edición codificada en el genoma única y detectable. facilitando el seguimiento de la presencia, intensidad, duración y momento relativo de estos eventos.
En bacterias, el registro transcripcional basado en una maquinaria de adquisición de espaciador CRISPR que contiene una RT permite la captura directa y cronológica de secuencias del ARN transcrito en el genoma. La aplicación de esta tecnología se ha ampliado a estudios in vivo del microbioma intestinal en ratones.
Aplicaciones terapéuticas ex vivo
La edición ex vivo del genoma basada en CRISPR de células derivadas de pacientes, seguida de su expansión y retrasplante, es una estrategia poderosa para tratar trastornos genéticos que se manifiestan en las células sanguíneas.
Un ejemplo de ello son los esfuerzos exitosos para desarrollar tratamientos para las hemoglobinopatías genéticas, el primero de los cuales ya ha sido aprobado en Europa, el Reino Unido y los Estados Unidos.
La anemia de células falciformes (SCD) y la β-talasemia dependiente de transfusiones (TDT) son el resultado de una variedad de mutaciones en la subunidad β de la hemoglobina (HBB).
Aunque estas mutaciones podrían, en principio, abordarse mediante la edición correctiva del genoma, reactivando la γ-globina fetal. (HBG), que normalmente es silenciada después del nacimiento por el represor transcripcional BCL11A, ofrece una solución más manejable. Esto se ha logrado mediante la alteración dirigida de un potenciador eritroide en el gen BCL11A en células madre hematopoyéticas CD34+ mediante electroporación Cas9 RNP.
Las terapias con células T con receptor de antígeno quimérico (CAR) implican la ingeniería ex vivo de células T para atacar marcadores de células cancerosas como CD19 o el antígeno de maduración de células B (BCMA) en neoplasias malignas de células B y mieloma múltiple, respectivamente, destruyendo así las células cancerosas después de la infusión en los pacientes.
En las terapias con células CAR-T autólogas actualmente aprobadas, el gen que codifica CAR se inserta aleatoriamente en el genoma mediante transducción con un vector lentiviral. Además, las células requieren un proceso de fabricación logísticamente complejo y propenso a fallos.
Para abordar estas limitaciones, las células CAR-T alogénicas generadas mediante la edición CRISPR de células T derivadas de donantes podrían ofrecer una funcionalidad mejorada, una producción optimizada y un control de calidad mejorado.
La inserción dirigida por CRISPR de la construcción CAR en el locus constante α del receptor de células T (TRAC) utilizando HDR evita la enfermedad de injerto contra huésped, mientras que las inactivaciones de los genes PDCD1, Regnase-1 y TGFBR2 están diseñadas para reducir el agotamiento de las células CAR-T y aumentar la persistencia.
Actualmente se están llevando a cabo varios ensayos clínicos para terapias con células CAR-T editadas por CRISPR que se dirigen a un panel de cánceres de la sangre. En un ejemplo notable, las células CAR-T editadas con bases multiplexadas se han utilizado para tratar la leucemia de células T recurrente, una afección previamente incurable, en pacientes infantiles.
Una estrategia potencialmente poderosa para futuras terapias genómicas combina la edición del genoma CRISPR ex vivo con tecnologías iPSC. Un enfoque desarrollado recientemente y diseñado para controlar la diabetes tipo 1 implica modificar el genoma de las iPSC, diferenciarlas en células del endodermo pancreático y encapsularlas en dispositivos biocompatibles para su implantación en pacientes para permitir la producción de insulina, que es vital para controlar la diabetes tipo 1.
Dentro de estos complejos protocolos, la edición CRISPR podría usarse para eliminar un conjunto de genes para promover la aceptación del injerto, proteger las células diseñadas del estrés y optimizar la producción de insulina.
La edición de genes CRISPR también se está utilizando en estudios de xenotrasplantes en animales. Por ejemplo, las modificaciones genéticas dirigidas que previenen el rechazo inmunológico en xenoinjertos de pulmón de cerdo han mejorado la aceptación del injerto en receptores de primates no humanos.
Aplicaciones terapéuticas in vivo
A diferencia de las estrategias ex vivo, la edición terapéutica de genes in vivo implica la administración directa de editores del genoma CRISPR a los tejidos afectados del cuerpo mediante administración localizada o sistémica.
Varios estudios preclínicos de prueba de principio ya han demostrado con éxito la edición terapéutica del genoma in vivo, ejemplificado notablemente por los esfuerzos para restaurar la expresión de la proteína distrofina en modelos animales de distrofia muscular de Duchenne mediante la administración dirigida a los músculos de Cas9 y ARN guía utilizando vectores AAV9. y rescatar la atrofia muscular espinal en un modelo de ratón de la enfermedad mediante la administración de CBE al cerebro mediada por AAV9.
Otro enfoque temprano implica la inyección directa de componentes CRISPR en el ojo para mitigar la amaurosis congénita de Leber tipo 10, una distrofia retiniana causada por mutaciones patógenas en el gen CEP290.
En este caso, Cas9, impulsado por promotores específicos de tejido para mayor precisión, se empaquetó junto con ARN guía dentro de vectores AAV5 y se entregó al área afectada mediante inyección subretiniana para restaurar la visión en ratones y primates no humanos.
El hígado es un objetivo excelente para la edición de genes in vivo debido a sus estructuras capilares discontinuas que facilitan la absorción de nanopartículas lipídicas administradas sistémicamente a través de la vía del receptor de lipoproteínas de baja densidad (LDL).
La amiloidosis por transtiretina (TTR) es una enfermedad causada por la acumulación dañina de TTR mal plegada en el corazón o el sistema nervioso. Como la TTR se produce predominantemente en el hígado, una estrategia terapéutica eficaz para la amiloidosis por TTR ha utilizado la administración mediada por LNP de ARNm de Cas9 y ARNsg sintéticos para desactivar este gen en el hígado. En un ensayo clínico, este enfoque demostró una reducción significativa de los niveles séricos de TTR, el primer caso exitoso de una terapia génica CRISPR in vivo administrada sistémicamente.
Los ensayos clínicos también están explorando tratamientos para la hipercolesterolemia familiar, una afección caracterizada por niveles elevados de colesterol LDL en sangre. Uno de los enfoques innovadores implica la administración sistémica basada en LNP de CRISPR ABE para atacar el gen de la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9) en el hígado, con el objetivo de reducir el colesterol LDL en sangre al eliminar este gen.
En primates no humanos, este enfoque ha disminuido significativamente los niveles de PCSK9 y el colesterol LDL sérico sin afectar los tejidos de la línea germinal, sentando las bases para los ensayos clínicos de fase I.
Una estrategia terapéutica mejorada utiliza la encapsulación de ARNm de ABE junto con ARNsg específicos de PCSK9 dentro de LNP que están formulados de forma única con ligandos multivalentes de N-acetilgalactosamina (GalNAc), lo que mejora la absorción por parte de las células hepáticas en pacientes con receptores de LDL deficientes.
El futuro: tecnologías emergentes
La búsqueda en curso de nuevas nucleasas guiadas por RNA está motivada en gran medida por la necesidad de enzimas alternativas con estructuras de RNA guía ortogonales y capacidades de direccionamiento, idealmente con un tamaño mínimo que facilite la construcción de proteínas de fusión (como BE y PE) y su eficiencia. Entrega celular utilizando vectores virales.
Esto se ejemplifica con el reciente desarrollo e ingeniería de la nucleasa Cas12f ultracompacta (422 aminoácidos) para mejorar su actividad de edición in vivo mediante escaneo mutacional profundo y diseño guiado por estructura.
El conjunto de herramientas de edición del genoma molecular también se ha ampliado recientemente gracias a los estudios en curso que tienen como objetivo descubrir nuevas nucleasas guiadas por RNA de origen natural. Estos esfuerzos se han centrado en los ancestros evolutivos de las enzimas Cas9 y Cas12, a saber, IscB/IsrB y TnpB, respectivamente.
Estas proteínas, también denominadas colectivamente nucleasas HEARO (RNA y ORF asociado a endonucleasa HNH) u OMEGA (actividad guiada por elemento móvil obligado), están codificadas dentro de elementos transponibles procarióticos en los que contribuyen al mecanismo de transposición y promueven la retención de transposones. Se ha demostrado que las nucleasas IscB y TnpB median en la edición programable del genoma en células humanas.
El descubrimiento de proteínas eucarióticas similares a TnpB denominadas Fanzors (~400–700 aminoácidos), que también se ha demostrado que catalizan el DNA guiado por RNA. escisión y soporte de edición programable del genoma, ha puesto de relieve la universalidad de las nucleasas guiadas por RNA en todos los ámbitos de la vida.
El tamaño compacto de estas nucleasas debería facilitar su administración; sin embargo, se necesitarán más esfuerzos de ingeniería molecular para aumentar su actualmente baja eficiencia y su limitado alcance de focalización.
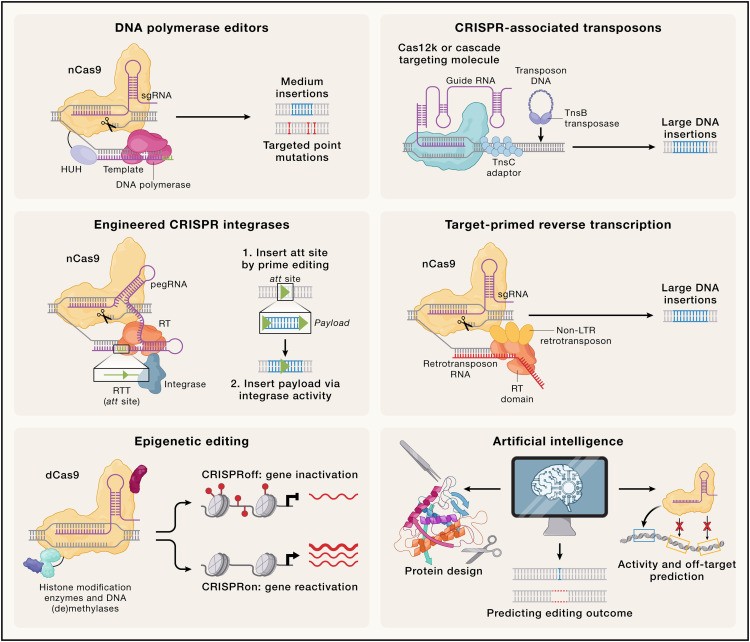
Editores de DNA polimerasa
A diferencia de los PE, en los que una RT se fusiona con una nickasa Cas9 para introducir modificaciones en la plantilla de RNA del sitio objetivo utilizando parte del RNA guía como plantilla, se están explorando enfoques novedosos que utilizan DNA polimerasas para introducir mutaciones específicas en el genoma.
En un primer intento, se logró una diversificación continua de nucleótidos dentro de una ventana ajustable de hasta 350 nucleótidos desde un sitio objetivo utilizando una DNA polimerasa diseñada y propensa a errores fusionada a una variante de nickasa Cas9.
Un estudio más reciente demostró que una DNA polimerasa derivada de fagos (proporcionada en trans) puede introducir ediciones en un sitio con mella de Cas9 utilizando una plantilla de DNA lineal atada.
A diferencia de la edición primaria basada en RT, este enfoque evita el emparejamiento de bases intramoleculares autoinhibitorias dentro del RNA guía y permite inserciones más largas de más de 100 nucleótidos.
Otro enfoque novedoso, denominado edición de clic, combina Cas9 con DNA polimerasas dependientes de DNA (DDP) y endonucleasas HUH para permitir la introducción de diversas ediciones del genoma, incluidas todas las sustituciones de un solo nucleótido, así como inserciones y eliminaciones breves.
El proceso aprovecha la actividad de bioconjugación de las endonucleasas HUH para unir covalentemente plantillas de «DNA de clic» a una fusión de proteínas HUH-nCas9-DDP. Este enfoque no sólo facilita la edición precisa del genoma con indeles mínimos sino que también evita inserciones no deseadas.
Las tecnologías de edición basadas en DNA polimerasa destacan por su potencial para inducir un amplio espectro de alteraciones genéticas, ofreciendo un alto grado de control y diversidad de resultados.
Recombinasas y transposones guiados por CRISPR
La edición del genoma que se basa en la reparación dirigida por plantillas de roturas del DNAbc se limita en gran medida a las células en proliferación en las que HDR está activo y no funciona de manera eficiente en las células posmitóticas.
Además, la eficiencia de las inserciones mediadas por HDR aumenta inversamente con el tamaño del inserto, lo que limita gravemente la capacidad de insertar fragmentos largos de DNA o genes completamente nuevos en el genoma.
Aunque la edición principal facilita la introducción de inserciones sin depender de HDR, actualmente las inserciones están restringidas a unas pocas decenas de nucleótidos.
Las capacidades de las PE se han ampliado recientemente mediante métodos que las combinan con serina recombinasas/integrasas, mediante los cuales la instalación mediada por el editor principal de un sitio de reconocimiento de recombinasa permite la inserción posterior de secuencias de DNA de múltiples kilobases por parte de la recombinasa. de nuevas recombinasas facilitará una mayor optimización de estos métodos para mejorar su eficiencia y especificidad.
Los transposones son capaces de catalizar de forma autónoma la inserción de grandes extensiones de DNA independientemente de la generación y reparación de DSB. Varios estudios han intentado fusionar Cas9 nucleasa inactiva con varias transposasas, incluidas La Bella Durmiente, Mariner y PiggyBac, para la transposición mediada por RNA en las células.
Aunque estos enfoques mejoran la frecuencia de los eventos de transposición en las proximidades del sitio objetivo. , la baja eficiencia general y la alta frecuencia de eventos de transposición fuera del objetivo actualmente impiden su uso generalizado como tecnologías de inserción genética dirigida.
A diferencia de las fusiones de transposasa Cas9 diseñadas, los sistemas de transposones asociados a CRISPR (CAST) son elementos transponibles similares a Tn7 de origen natural que han adoptado sistemas CRISPR-Cas de tipo I o V como módulos de orientación para mediar la transposición de DNA guiada por RNA. Los sistemas son capaces de una inserción altamente eficiente y específica en un sitio en bacterias.
Amplios estudios estructurales y funcionales han arrojado luz sobre los mecanismos de los CAST, proporcionando un marco para la ingeniería molecular de estos sistemas para permitir la transposición de DNA dirigida en bacterias. células de mamíferos.
Aunque la eficiencia de los CAST de tipo V sigue siendo bastante baja, los CAST de tipo I muestran signos prometedores de actividad en células humanas, lo que destaca el potencial de estos sistemas como tecnologías para la inserción específica de un sitio de grandes cargas genéticas.
Sin embargo, será necesario realizar más perfiles funcionales sistemáticos de los CAST naturales, junto con investigaciones mecanicistas y esfuerzos de ingeniería, para alcanzar los niveles de eficacia necesarios para una edición sólida del genoma y la administración terapéutica de genes.
Edición basada en retroelementos
Edición del epigenoma
La introducción de modificaciones genéticas permanentes plantea riesgos importantes, como mutaciones no deseadas, y plantea preocupaciones éticas en el contexto de la edición del genoma de la línea germinal.
La edición del epigenoma basada en CRISPR, que introduce modificaciones epigenéticas específicas que no afectan el ADN de la línea germinal pero que pueden persistir a lo largo de numerosas generaciones celulares, ofrece una alternativa prometedora.
Al fusionar enzimas modificadoras de ADN e histonas con Cas9 sin nucleasas, la cromatina se puede reestructurar en un locus genómico específico para inducir o reprimir la expresión del gen objetivo. Un enfoque de editor de epigenomas recientemente establecido denominado CRISPRoff explota las DNA metiltransferasas como Dnmt3A o Dnmt3L junto con dominios represores transcripcionales KRAB para silenciar de manera eficiente y persistente la expresión génica en células en proliferación a lo largo de múltiples generaciones.
El silenciamiento puede revertirse mediante CRISPRon, un editor multipartito que combina Cas9 con la DNA desmetilasa TET1 y dominios de activación transcripcional, para reactivar la expresión. La acetilación es otra forma de modificación epigenética de histonas en la que las histonas acetiltransferasas (HAT) y las histonas desacetilasas (HDAC) se pueden utilizar para modificar la cromatina y así activar o reprimir la expresión génica, respectivamente.
Sin embargo, persisten ciertas limitaciones. Como Cas9 puede unirse transitoriamente a sitios fuera del objetivo basándose únicamente en la secuencia semilla del RNA guía, los modificadores de histonas y cromatina pueden afectar inadvertidamente el estado de transcripción de genes fuera del objetivo.
Además, los efectos a largo plazo y la estabilidad de estos cambios epigenéticos aún deben investigarse con más detalle para comprender completamente sus consecuencias, especialmente en el contexto de enfermedades complejas.
Edición de RNA
Al igual que con la edición del epigenoma, la introducción de mutaciones dirigidas directamente en el mRNA ofrece una alternativa potencialmente más segura a la edición del DNA debido a la vida útil limitada de los mRNA dentro de la célula. Los enfoques de edición de RNA han evolucionado para ofrecer un control más preciso y versátil sobre los resultados de la edición.
Uno de esos avances recientes surgió del descubrimiento del sistema Cas7-11, un novedoso efector CRISPR-Cas de una sola proteína que utiliza RNA guía para escindir los RNA objetivo.
El sistema ha sido rediseñado para anular transcripciones con una mínima actividad fuera del objetivo y aparentemente no hay actividad de escisión colateral en células de mamíferos, en comparación con los enfoques de eliminación mediada por Cas13a.
Además, recientemente se ha informado de un enfoque novedoso para el trans-empalme mediado por CRISPR de transcripciones de RNA largas.
Al utilizar enzimas Cas13 dirigidas a RNA en combinación con un RNA de transempalme, la tecnología permite el reemplazo o la inserción de segmentos grandes en transcripciones de mRNA de mamíferos.
Estas nuevas tecnologías tienen implicaciones importantes para el desarrollo terapéutico, particularmente para enfermedades genéticas que podrían abordarse mediante la corrección genética transitoria. Por lo tanto, la edición de RNA representa una oportunidad para enfoques más sofisticados y específicos en medicina genómica y para ampliar el potencial de tratamientos que requieren modulación genética transitoria o reversible.
Nuevos métodos de entrega
La entrega eficiente y precisa de los complejos del editor de genes en células u órganos diana sigue siendo el factor limitante más importante para la edición del genoma. Actualmente se están desarrollando varias estrategias para mejorar la eficiencia de la entrega y minimizar la posible inmunogenicidad de los editores de genoma basados en CRISPR.
En primer lugar, recientemente se han desarrollado nuevas formulaciones de LNP para permitir la focalización en tejidos específicos, ofreciendo así un enfoque versátil para empaquetar y administrar componentes CRISPR con una mayor absorción celular y una reducción de la edición fuera del objetivo.
Los péptidos que penetran en las células también han mostrado un potencial considerable como método de administración de enzimas CRISPR, particularmente en la edición de linfocitos humanos primarios, células neuronales y epitelios de las vías respiratorias.
Los sistemas de inyección contráctil bacteriana se han rediseñado para permitir la administración transitoria y específica de la célula de carga de proteínas, incluidas las nucleasas editoras del genoma, con alta eficiencia. Las partículas similares a virus diseñadas que imitan a los virus pero que carecen de material genético viral han surgido como una poderosa alternativa a los vectores virales para aplicaciones in vivo.
Otra tecnología prometedora basada en la ingeniería de partículas virales endógenas se basa en virus antiguos codificados en genomas eucariotas, que podrían permitir la administración con una respuesta inmunogénica reducida. Finalmente, con la llegada de enfoques precisos de diseño computacional de proteínas, se están desarrollando jaulas de proteínas diseñadas de novo que podrían proporcionar estrategias modulares para entregar complejos editores de genes a entornos biológicos que no son accesibles a los sistemas naturales, a pesar de la gran promesa que encierran estos avances.
Sin embargo, será necesario evaluar rigurosamente los perfiles de citotoxicidad, fuera del objetivo e inmunogenicidad de estas tecnologías en varios tipos de células y tejidos.
Además, la escalabilidad de algunos de estos métodos para aplicaciones clínicas plantea un desafío importante, al igual que el costo de desarrollar y fabricar sistemas de administración tan complejos. En general, existe una necesidad crítica de realizar más investigaciones para perfeccionar estas técnicas y garantizar su seguridad y eficacia para uso terapéutico.
Inteligencia artificial
La reciente y rápida adopción de la inteligencia artificial (IA) y los algoritmos computacionales de aprendizaje profundo en las ciencias biológicas también ha tenido implicaciones de gran alcance para el campo de la edición del genoma.
Ya ha logrado avances considerables en la mejora de su precisión y eficiencia mediante el modelado predictivo de los resultados y la eficiencia de las ediciones, incluida la actividad fuera del objetivo, que son cruciales para minimizar alteraciones genéticas no deseadas, particularmente para aplicaciones terapéuticas donde la seguridad y la eficacia son de suma importancia.
Potencialmente, los métodos de IA podrían usarse en el futuro para personalizar aún más los enfoques de edición de genes en aplicaciones terapéuticas.
El poder de la IA también se está aprovechando para la ingeniería de nucleasas novedosas, más eficientes y específicas a través de novedosos métodos de diseño computacional de proteínas. Sin embargo, los métodos de IA también conllevan su propio conjunto de desafíos. Los modelos predictivos dependen en gran medida de la disponibilidad y calidad de los datos de entrenamiento, que pueden ser limitados y muy variables según el enfoque experimental.
Además, la naturaleza de «caja negra» de los modelos de IA impide comprender cómo estos modelos toman decisiones, lo que hace que las predicciones sean difíciles de interpretar y de confiar. Sin embargo, las aplicaciones potenciales de la IA en la edición de genes son inmensas, pero requerirán una cuidadosa consideración en el futuro.
Panorama
En la última década, el campo de la edición del genoma ha pasado de ser una actividad científica incipiente a una fuerza biotecnológica transformadora.
Las tecnologías CRISPR de primera generación, basadas principalmente en reparación endógena o DSB de sitio específico, se han complementado con tecnologías de segunda generación, como la edición base y la edición principal, que permiten la modificación directa del DNA objetivo sin generación de DSB y, en general, se consideran más seguras y producen resultados más predecibles.
Se están desarrollando tecnologías CRISPR emergentes (de tercera generación) para abordar dos necesidades importantes no satisfechas en este campo: lograr la inserción precisa de cargas útiles grandes (del tamaño de un gen) y la regulación genética sin ninguna edición del genoma mediante ingeniería del epigenoma.
El avance continuo de estas y otras tecnologías es posible gracias a dos enfoques: por un lado, mediante la extracción del metagenoma para descubrir nuevos sistemas moleculares y, por el otro, mediante la biología sintética y la ingeniería molecular (apoyada por la IA).
El mejor ejemplo del estado actual de la edición del genoma es un repertorio en constante expansión de tecnologías y métodos novedosos, basados principalmente en herramientas moleculares guiadas por RNA derivadas de sistemas CRISPR-Cas, que han revolucionado nuestra capacidad para manipular el material genético con precisión y facilidad.
La trayectoria actual sugiere que estas tecnologías seguirán perfeccionándose, con mejoras graduales en la especificidad, la eficiencia y los mecanismos de entrega. Las áreas que justifican una mayor investigación incluirán el desarrollo de vectores de administración más específicos y menos inmunogénicos, la reducción de efectos fuera del objetivo y un mayor control sobre los resultados de la edición, mejorando tanto la exactitud como la precisión de la edición del genoma.
De cara al futuro, es probable que los sistemas guiados por ácidos nucleicos sigan siendo fundamentales para la edición del genoma debido a su simple programabilidad y adaptabilidad, en comparación con las tecnologías basadas puramente en proteínas.
Sin embargo, el modo de su aplicación puede evolucionar, con posibles cambios hacia enfoques de edición más transitorios para minimizar el riesgo de cambios genéticos no deseados a largo plazo o entrega transitoria para minimizar la alteración genómica y la respuesta inmune.
Dado que las tecnologías de primera generación ya han sido aprobadas para uso clínico en el tratamiento de afecciones como la ECF y la β-talasemia mediante edición ex vivo, y los enfoques in vivo no se quedan atrás, el cuello de botella que limita el desarrollo de futuras terapias dirigidas por el genoma para una amplia gama de Las enfermedades ya no serán necesariamente la falta de editores genómicos seguros, eficientes y precisos. Los principales desafíos residirán en los métodos de administración, especialmente para órganos distintos de la sangre y el hígado y para ciertos tipos de células ex vivo.
Es probable que los avances en los métodos de administración in vivo, como los observados en el desarrollo de vacunas de mRNA, catalicen significativamente la aplicación de tecnologías CRISPR. Además, incluso con los tejidos y células que actualmente se pueden editar utilizando los métodos disponibles, se necesitará más investigación básica para identificar objetivos de edición seguros.
La gama de enfermedades que podrían tratarse mediante CRISPR se ampliará con el desarrollo de nuevos vectores de administración y la caracterización de variantes causantes de enfermedades y sus estrategias correctivas. Se espera que los avances fuera del campo CRISPR ayuden a impulsar las tecnologías en nuevas direcciones, mejorando sus capacidades.
En este contexto, el auge de los métodos basados en IA nos permitirá modelar con precisión paisajes complejos de edición genómica, predecir resultados de edición dentro y fuera del objetivo y diseñar editores de genoma más capaces, acelerando así el ritmo de implementación de enfoques terapéuticos seguros.
Las implicaciones éticas y sociales, en particular las relativas a las modificaciones del genoma de la línea germinal humana, seguirán siendo un tema central en los debates sobre la edición del genoma.
Ahora que la edición de células somáticas humanas se está haciendo realidad, la perspectiva de una edición terapéutica y no terapéutica de la línea germinal, con el potencial de realizar cambios hereditarios en el genoma humano, plantea profundas cuestiones éticas que la comunidad global debe abordar. Como han demostrado los estudios de investigación en embriones humanos, las tecnologías de edición del genoma CRISPR no son lo suficientemente seguras o eficaces como para utilizarlas en la edición de la línea germinal con fines reproductivos.
Además, la utilidad terapéutica de la edición de la línea germinal es limitada y probablemente beneficie solo a un pequeño número de personas. Sin embargo, no se puede subestimar la urgencia de lograr un consenso internacional sobre la gobernanza y la administración responsable de las tecnologías de edición del genoma, especialmente a la luz de su rápido desarrollo, mejora continua y adopción generalizada.
En conclusión, a pesar de los desafíos existentes, el futuro de la edición del genoma CRISPR es brillante. Tiene el potencial no sólo de impulsar avances en la investigación y revolucionar la medicina humana, sino también de mejorar la agricultura y abordar los desafíos ecológicos, sentando así las bases para un futuro más saludable y sostenible para las generaciones venideras.
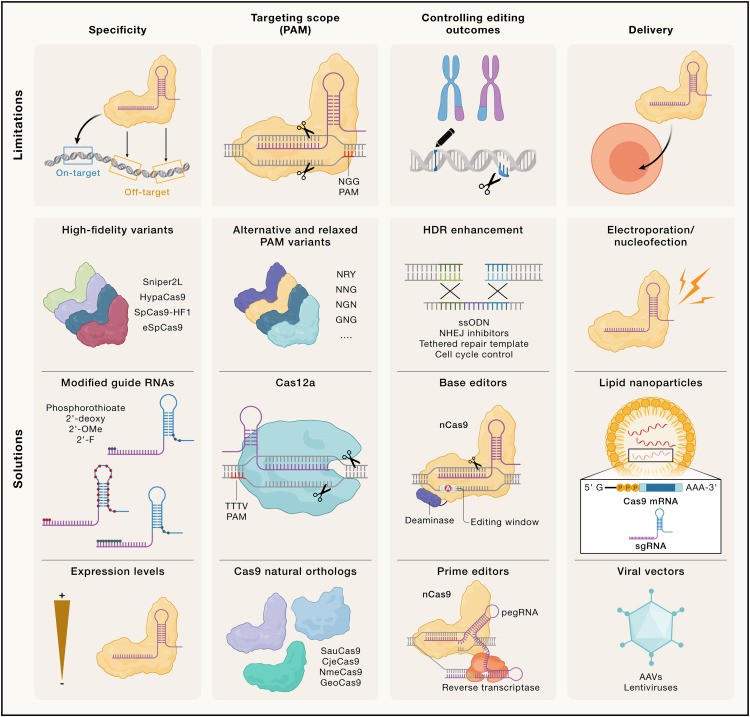
Figura 3 Tecnologías de edición CRISPR actuales
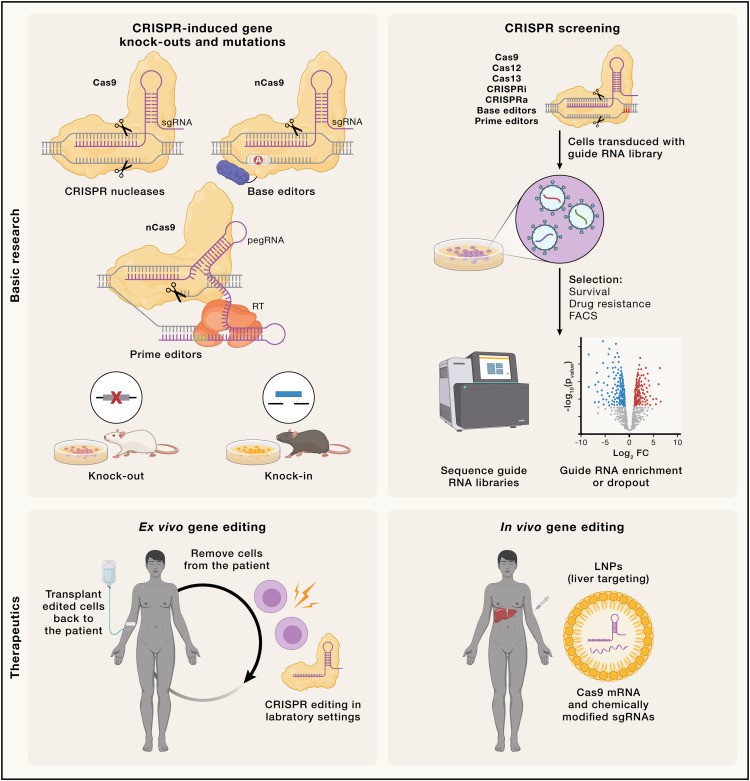
Figura 4
Figura 5. Tecnologías emergentes en la edición del genoma
Nucleasas ancestrales mínimas guiadas por RNA