Ronald Palacios Castrillo
La atención de los pacientes con amiloidosis sistémica de cadenas ligeras de inmunoglobulinas (AL) ha experimentado cambios transformadores, lo que ha llevado a un progreso marcado y constante en los resultados de los pacientes durante las últimas cuatro décadas.
Se han logrado avances sustanciales mediante la implementación de tratamientos dirigidos a la discrasia de células plasmáticas subyacente, en su mayoría adaptados del tratamiento del mieloma. En la última década se han producido avances notables que han infundido esperanza entre los pacientes con amiloidosis AL.
Esta revisión se centra en los avances recientes en nuestra comprensión de la patogénesis y las características clínicas de la fibrilogénesis amiloide, así como en la estratificación del riesgo y los avances terapéuticos, y describe las necesidades no cubiertas en la amiloidosis AL.
=> Recibir por Whatsapp las noticias destacadas
La amiloidosis comprende un grupo de enfermedades desencadenadas por el mal plegamiento de una proteína precursora soluble. Este mal plegamiento conduce a la formación de oligómeros, agregados y fibrillas de amiloide caracterizadas por láminas β plisadas, que se depositan extracelularmente en diversos órganos y tejidos. El resultado es una disfunción orgánica progresiva, insuficiencia orgánica y, finalmente, la muerte.1.
La disfunción orgánica se debe a la alteración de la arquitectura causada por depósitos de amiloide, efectos citotóxicos directos de agregados u oligómeros de proteínas, o ambos.2.
Hasta la fecha se han identificado un total de 42 proteínas amiloidogénicas precursoras solubles que pueden formar fibrillas de amiloide extracelulares.3.
Las amiloidosis se clasifican en sistémicas o localizadas y se clasifican además según el sitio de los depósitos de amiloide y el sitio de producción de la proteína precursora. La amiloidosis sistémica puede ser hereditaria o adquirida. Las dos formas más comunes, la amiloidosis AL y la amiloidosis por transtiretina de tipo nativo (ATTRwt), son adquiridas. Aunque ambas formas de amiloidosis sistémica son comunes, la amiloidosis ATTRwt es más prevalente.
La amiloidosis AL se asocia con una discrasia clonal de células plasmáticas y es causada por una producción anormal o excesiva de cadenas ligeras de inmunoglobulina amiloidogénica, que se agregan en oligómeros y fibrillas de amiloide, lo que conduce a una disfunción orgánica.4. Amiloidosis ATTRwt, que afecta a hombres mayores de 70 años, es causada por la agregación de transtiretina normal (TTR) y produce predominantemente miocardiopatía.
La amiloidosis sistémica hereditaria o familiar también puede ser causada por mutaciones genéticas heredadas de forma autosómica dominante.5. Más de 120 mutaciones puntuales en el gen que codifica TTR, una proteína transportadora de tiroxina y proteína fijadora de retinol, pueden causar amiloidosis sistémica que afecta principalmente a los sistemas nerviosos periférico y autónomo y el corazón.6.
Patogénesis
Una característica patognomónica de la amiloidosis sistémica es el plegamiento anormal de una proteína precursora soluble normal (Figura 1).
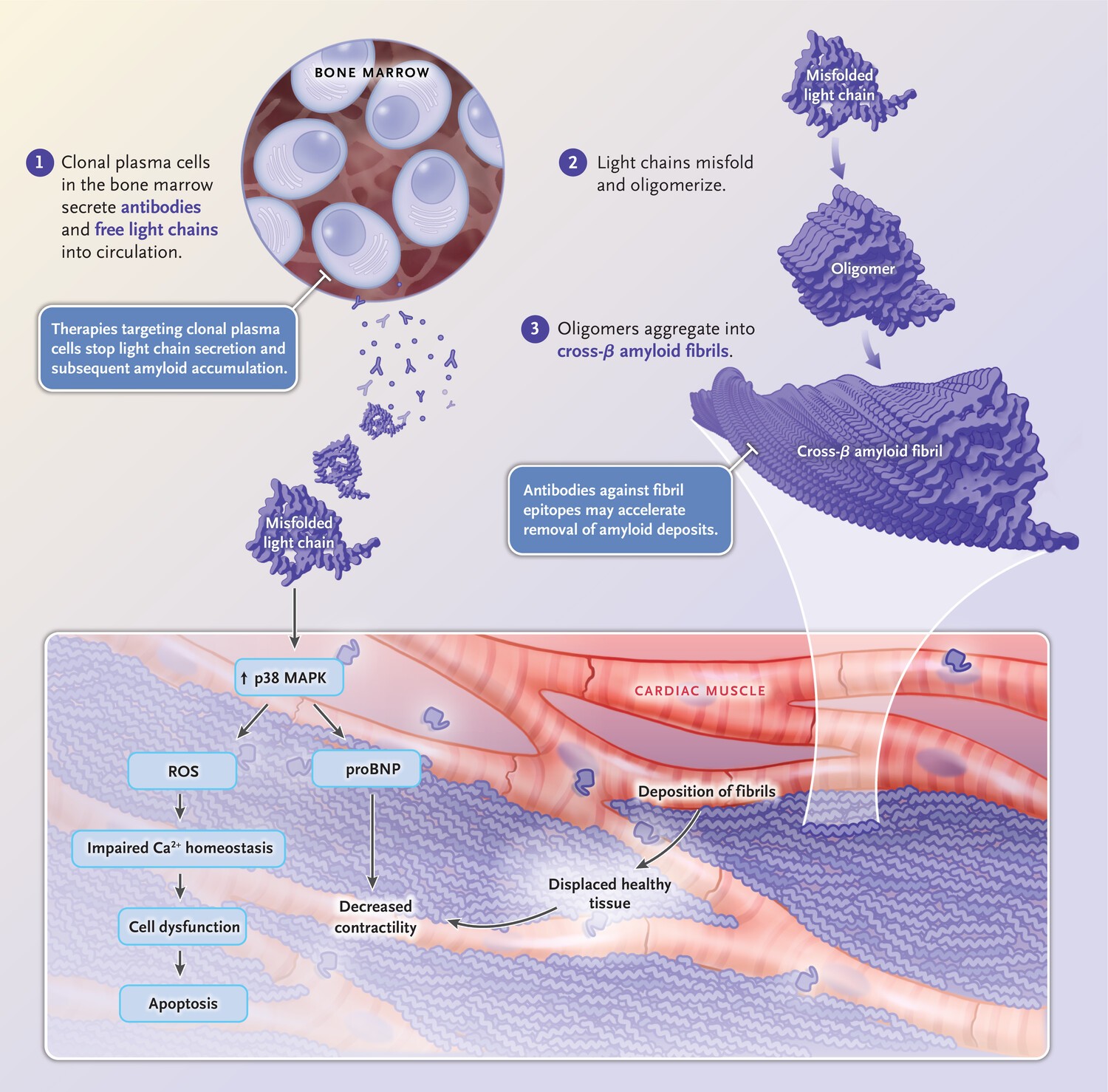
En la amiloidosis AL, el plegamiento anormal es el resultado de un evento proteolítico o de una secuencia de aminoácidos que hace que una cadena ligera de inmunoglobulina sea termodinámica y cinéticamente inestable, lo que lleva a la autoagregación.7.
Estos agregados interactúan con el glicosaminoglicano y la proteína P amiloide sérica, promoviendo formación de fibrillas y estabilización de los depósitos de amiloide en los tejidos, alterando la arquitectura del tejido y, en última instancia, provocando disfunción orgánica.
La evidencia emergente de los modelos de Caenorhabditis elegans(8) y pez cebra(9) sugiere que los agregados de precursores amiloidogénicos también tienen efectos citotóxicos directos que contribuyen a la disfunción orgánica.
La proteostasis, un mecanismo celular, normalmente garantiza el plegamiento y la función adecuados de las proteínas.10. Sin embargo, la mutación genética, la proteostasis alterada debido al envejecimiento u otros factores pueden favorecer el plegamiento incorrecto y la agregación.
Los agregados de proteínas forman fibrillas de amiloide, caracterizadas por estructuras de fibras β cruzadas no paralelas. Las fibrillas de amiloide tienen un diámetro de 8,0 a 10,0 nm, según se determina mediante microscopía electrónica.11.
Puntos clave
- Amiloidosis sistémica de cadenas ligeras
- La amiloidosis de cadenas ligeras de inmunoglobulinas (AL) es una enfermedad rara que ocurre cuando un trastorno de las células plasmáticas produce cadenas ligeras monoclonales, que se pliegan mal y se depositan como fibrillas en órganos o tejidos.
- Los signos y síntomas, el enfoque del tratamiento y el pronóstico son muy variables y están dictados por la naturaleza de las cadenas ligeras únicas de cada paciente.
- Uno de los determinantes más importantes de la supervivencia es la gravedad de la afectación cardíaca.
- El arsenal de terapias para la amiloidosis AL se está expandiendo rápidamente y ofrece una perspectiva prometedora para los pacientes en 2024.
- La supervivencia general ha mejorado considerablemente, pero persisten muchas necesidades insatisfechas.
La amiloidosis AL se asocia típicamente con un trastorno de las células plasmáticas que es responsable de producir cadenas ligeras de inmunoglobulina lambda en el 75 al 80 % de los casos y cadenas ligeras kappa en el 20 al 25 % restante.
La amiloidosis AH, resultante de cadenas pesadas de inmunoglobulinas, y la amiloidosis AH/AL, resultante de cadenas pesadas y ligeras de inmunoglobulinas, son mucho menos comunes.12.
La translocación cromosómica t(11;14), que reúne el locus de cadenas pesadas de inmunoglobulinas (IgH ) y el oncogén ciclina D1, es característico de la amiloidosis AL y ocurre en aproximadamente el 50% de los casos(13), mientras que la hiperdiploidía, que es común en el mieloma, se observa en aproximadamente el 10% de los casos de amiloidosis AL.14.
Mutaciones somáticas en el grupo IGLV de genes que codifican la región variable de la cadena ligera disminuyen la estabilidad de las proteínas, lo que facilita la formación de fibrillas de amiloide.15.
Factores de riesgo
Los factores de riesgo de amiloidosis AL aún no están claros, pero son comunes la gammapatía monoclonal y el mieloma preexistentes. Entre los pacientes con gammapatía monoclonal de origen desconocido (GMSI), el riesgo relativo es de 8,8,16 con una incidencia del 1 % de amiloidosis AL observada en un estudio que incluyó a 1384 pacientes con GMSI.
La amiloidosis AL se diagnostica hasta en 10 a 15% de los pacientes con mieloma, y 38% de los pacientes con mieloma tienen depósitos de rojo Congo positivos en aspirados de grasa subcutánea, muestras de biopsia de médula ósea o ambas.17. Un aumento en el suero monoclonal de Los niveles de cadenas ligeras libres preceden el desarrollo de amiloidosis AL en más de 4 años en todos los pacientes.18.
La exposición al Agente Naranja, un herbicida utilizado en la Guerra de Vietnam, puede estar asociada con la amiloidosis AL, aunque la evidencia es limitada.19. N-glicosilación de cadenas ligeras kappa monoclonales puede servir como factor predictivo para un diagnóstico más temprano de amiloidosis AL en pacientes con GMSI(20,21).
Epidemiología
Los datos epidemiológicos sobre la amiloidosis AL son limitados, principalmente debido a la ausencia de bases de datos poblacionales completas. La prevalencia de esta enfermedad tiende a aumentar con la edad.
En el Proyecto del Condado de Olmsted en Minnesota, la tasa de incidencia general de amiloidosis AL fue de 8,9 casos por 1 millón de personas-año entre 1950 y 1989, 10,5 casos por 1 millón de personas-año entre 1970 y 1989, y 12,0 casos por 1 millón de personas-año entre 1990 y 2015(22). Se informó una tasa bruta de incidencia calculada de 10,4 casos por 1 millón de personas-año en 38 países.
En 2018, se habían diagnosticado aproximadamente 74.000 casos de amiloidosis AL en todo el mundo en los 20 años anteriores.
La incidencia estimada fue de 10 casos por 1 millón de habitantes y la prevalencia estimada a 20 años fue de 51 casos por 1 millón de habitantes.23.
Un estudio del mundo real basado en una base de datos de reclamaciones de atención médica de EE. UU. mostró un aumento significativo en la prevalencia de amiloidosis AL, de 15,5 casos por millón de habitantes en 2007 a 40,5 casos por millón de habitantes en 2015, mientras que la tasa de incidencia se mantuvo estable, oscilando entre 9,7 y 14,0 casos por millón de personas-año.24.
Presentaciones clínicas
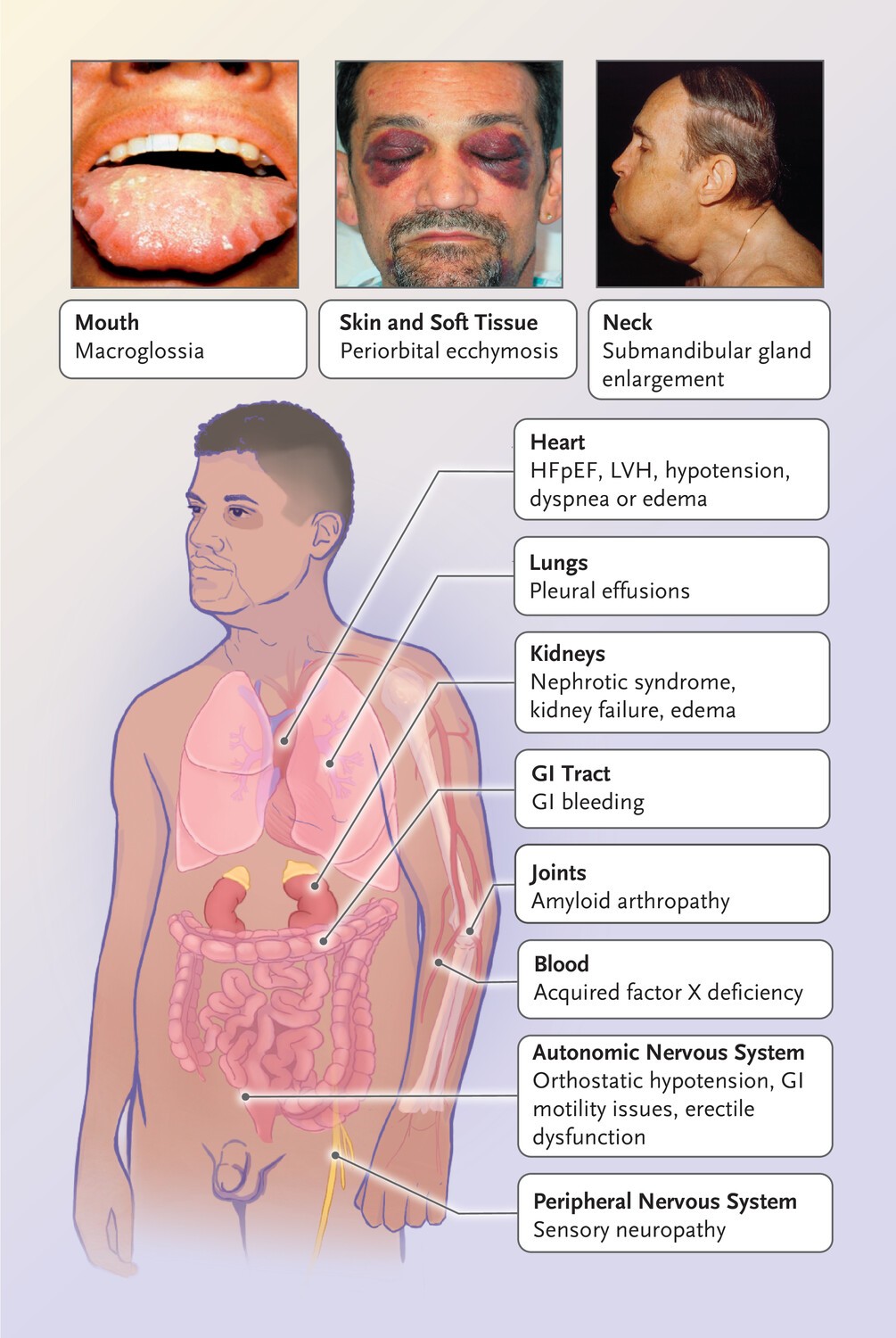
En la mayoría de los casos, la amiloidosis AL se caracteriza por ser una enfermedad rápidamente progresiva con diversos síndromes clínicos (Figura 2).
Los síntomas inespecíficos comunes incluyen fatiga y pérdida de peso; sin embargo, los síntomas específicos de un órgano a menudo conducen al diagnóstico. Los retrasos en el diagnóstico se deben a la falta de conocimiento entre los médicos.25.
Los riñones suelen estar afectados en la amiloidosis AL (en 60 a 70% de los pacientes); Los efectos renales se manifiestan típicamente como proteinuria en rango nefrótico, hipoalbuminemia, hiperlipidemia secundaria y edema. En algunos casos, la insuficiencia renal ocurre en ausencia de proteinuria, como resultado del depósito de amiloide intersticial o vascular. 26.
El corazón también se ve afectado con frecuencia (en 70 a 80% de los pacientes), y la afectación cardíaca es la principal causa de muerte. Los primeros signos incluyen bajo voltaje en la electrocardiografía y engrosamiento ventricular concéntrico en la ecocardiografía, junto con disfunción diastólica.
La contractilidad auricular deficiente se produce incluso en ritmo sinusal, y los pacientes con amiloidosis AL cardíaca tienen riesgo de desarrollar trombos auriculares y complicaciones tromboembólicas.
Son comunes los niveles elevados de troponina cardíaca sérica, propéptido natriurético tipo B N-terminal (NT-proBNP), o ambos. 27. Las bradiarritmias a menudo preceden a la descompensación cardíaca terminal.28.0
Los síntomas del sistema nervioso incluyen neuropatía de fibras pequeñas y disfunción autonómica, que se manifiesta como alteraciones de la motilidad gastrointestinal, saciedad temprana, ojos y boca secos, hipotensión ortostática y vejiga neurogénica.
La macroglosia se observa en aproximadamente del 10 al 20% de los pacientes. La afectación del hígado causa colestasis y hepatomegalia. La hiperbilirrubinemia puede ocurrir como un evento terminal en pacientes con afectación hepática. La afectación esplénica se manifiesta como hipoesplenismo funcional más que como esplenomegalia.
Pueden aparecer “moretones fáciles” como resultado de depósitos de amiloide o deficiencia del factor X de coagulación. También pueden ocurrir equimosis cutáneas, distrofia ungueal, alopecia y artropatía amiloide.
La presencia de una enfermedad multisistémica que no se explica por enfermedades comunes o fatiga general junto con cualquiera de estos síndromes clínicos debe impulsar la realización de pruebas de amiloidosis.
Los mecanismos precisos que rigen el tropismo orgánico en la amiloidosis AL aún no están claros. Ciertas características de los genes de la región variable de la cadena ligera elevan el riesgo de afectación de órganos específicos.
Por ejemplo, el gen de la línea germinal IGLV6-57 es más prevalente entre pacientes con manifestaciones renales, mientras que IGLV1-44 se asocia con un mayor riesgo de afectación cardíaca. La mayoría de los casos de amiloidosis AL sistémica se atribuyen a las cadenas ligeras lambda, pero la cadena ligera kappa de la mutación de la línea germinal IGKV1-33 se dirige al hígado(29,30).
Diagnóstico
Los síntomas inespecíficos relacionados con la amiloidosis AL a menudo contribuyen a retrasos en el diagnóstico. La consideración de la amiloidosis AL es crucial en pacientes con proteinuria inexplicable, miocardiopatía restrictiva, neuropatía periférica con características autonómicas, síndrome del túnel carpiano en ambas muñecas o hepatomegalia sin anomalías de imagen y en cualquier paciente con gammapatía monoclonal o mieloma múltiple con manifestaciones atípicas como macroglosia o ojos de mapache. Un alto índice de sospecha es fundamental para evitar retrasos en el diagnóstico.
El diagnóstico de amiloidosis AL requiere evidencia de depósitos de amiloide en el tejido (objetivo o sustituto) y evidencia de discrasia de células plasmáticas.
Los depósitos de amiloide tisular muestran birrefringencia verde cuando se tiñen con tinte rojo Congo y se observan con el uso de microscopía de luz polarizada.
La aspiración con aguja fina de grasa abdominal es un procedimiento simple que resulta positivo para los depósitos de amiloide en aproximadamente el 70 al 75% de los pacientes con amiloidosis AL .31.
Otros tejidos que permiten procedimientos de biopsia relativamente no invasivos son las glándulas salivales menores, la encía, el recto y piel. Sin embargo, si el índice clínico de sospecha es alto y la aspiración de la grasa abdominal es negativa para la tinción con rojo Congo, puede ser necesaria una biopsia de un órgano afectado para establecer el diagnóstico de amiloidosis.
El examen de muestras de biopsias de grasa abdominal y de médula ósea identifica al 85% de los pacientes con amiloidosis AL. 31.
Después de que se haya establecido un diagnóstico tisular de amiloidosis mediante biopsia de un órgano sustituto o diana, la confirmación de amiloidosis AL requiere la demostración de una discrasia de células plasmáticas de acuerdo con la presencia de una proteína monoclonal determinada mediante electroforesis de inmunofijación en suero u orina, libre de inmunoglobulinas ensayo de cadena ligera, la presencia de células plasmáticas restringidas lambda o kappa en una muestra de biopsia de médula ósea, o los tres.
La electroforesis por inmunofijación debe realizarse en muestras de suero y orina porque en la amiloidosis AL, a diferencia del mieloma múltiple, la concentración del componente monoclonal suele ser demasiado baja para detectarse mediante electroforesis de proteínas.
Incluso si se identifica una cadena ligera de inmunoglobulina monoclonal en suero u orina, la aspiración y biopsia de médula ósea son obligatorias para evaluar la carga de células plasmáticas y descartar mieloma múltiple y otros trastornos menos comunes que pueden asociarse con la amiloidosis AL, incluida los trastornos linfoproliferativos de células B como la leucemia linfocítica crónica, el linfoma indolente y la macroglobulinemia de Waldenström. 32.
Si se detectan depósitos de amiloide en muestras de biopsia, la identificación precisa de la proteína precursora es crucial para guiar el tratamiento.
Dicha identificación es factible con el uso de estudios inmunohistoquímicos (33), en laboratorios altamente experimentados y con el uso de microscopía electrónica con inmunooro. 34.
La precisión de los estudios inmunohistoquímicos depende no solo de la experiencia del laboratorio sino también de un amplio panel de anticuerpos para informar.
Éste no es el método de elección para tipificar con precisión las fibrillas de amiloide, aunque la tinción por inmunofluorescencia de una muestra de biopsia de riñón puede tener precisión suficiente para establecer un diagnóstico tisular de amiloidosis. Sin embargo, un análisis basado en espectrometría de masas de los tejidos que contienen amiloide se considera ahora el mejor enfoque, con una sensibilidad reportada del 88% y una especificidad del 96%(35,36).
Aunque no está ampliamente disponible, la espectrometría de masas se realiza en algunas laboratorios de referencias (clinica Mayo) para confirmar inequívocamente la subunidad proteica.
Es particularmente importante distinguir entre amiloidosis AL y amiloidosis variante V122I ATTR, especialmente en pacientes de raza negra, debido a la alta prevalencia de la forma variante y una presentación clínica que puede parecerse a la amiloidosis AL. Ambas afecciones pueden implicar gammapatía monoclonal, por lo que el diagnóstico y tratamiento precisos son esenciales para un tratamiento adecuado. 37.
Las imágenes cardíacas son un componente crítico de una evaluación cardíaca integral en pacientes con amiloidosis AL(38,39). La ecocardiografía (específicamente, las técnicas de imágenes de tensión y Doppler) ayudan a identificar los signos tempranos de amiloidosis cardíaca, como los patrones de llenado ventricular restrictivo.
La resonancia magnética cardíaca aporta información valiosa sobre el espesor del miocardio, el realce tardío con gadolinio y el mapeo ponderado en T1 del volumen extracelular.
La tomografía por emisión de positrones con el uso de radiotrazadores como el 18F-florbetapir apunta a los depósitos de amiloide, especialmente en el miocardio. Por el contrario, la gammagrafía ósea puede ser útil para diagnosticar la amiloidosis cardíaca ATTR. La integración de estas técnicas de imagen avanzadas permite una comprensión más matizada de la afectación cardíaca en la amiloidosis AL.
Sistema de estadificación y estratificación del riesgo
La supervivencia de los pacientes con amiloidosis AL sistémica depende de la gravedad de la disfunción cardíaca en el momento del diagnóstico.
Los pacientes que reciben un diagnóstico en una etapa tardía del curso clínico del trastorno (cuando el daño cardíaco suele estar avanzado) tienen una mediana de supervivencia de 3 a 6 meses, mientras que los pacientes sin afectación cardíaca pueden sobrevivir durante muchos años.
Los sistemas de estadificación actuales para la estratificación del riesgo y el pronóstico utilizan los biomarcadores de discrasia de células plasmáticas y afectación cardíaca y renal .
El sistema de estadificación de Mayo Clinic de 2004 se basa en los niveles de NT-proBNP y troponinas cardíacas(40) y fue modificado por investigadores europeos para identificar y clasificar a los pacientes de muy alto riesgo, es decir, aquellos con un nivel de NT-proBNP superior a 8500 pg por mililitro(41).
Este sistema de estadificación cardíaca es el más utilizado para predecir la muerte prematura. El sistema se modificó en 2012 para incluir la carga clonal, evaluada como la diferencia entre las cadenas ligeras libres circulantes involucradas y no involucradas (dFLC, con un valor de corte de 180 mg por litro), que es un predictor de supervivencia(42).
The Mayo Clinic 2012 El sistema de estadificación predice la supervivencia tardía con mayor precisión que el sistema de estadificación de Mayo Clinic 2004, y el sistema de estadificación de Mayo Clinic 2004 con la modificación europea predice la muerte temprana con mayor precisión que el sistema de 2012.
En la era actual de tratamientos eficaces contra el clon de células plasmáticas en pacientes con amiloidosis AL, la dFLC parece tener menos pronóstico en estos sistemas de estadificación(43). Investigadores de la Universidad de Boston introdujeron un sistema de estadificación que incorpora BNP y troponina I, que también predice la supervivencia(44 ,45).
Los pacientes con amiloidosis AL que tienen un nivel de dFLC muy bajo (<50 mg por litro) tienen un resultado sustancialmente mejor, independientemente del estadio cardíaco, que aquellos con niveles más altos de dFLC(46–48).
También se ha desarrollado un sistema de estadificación renal que utiliza biomarcadores de la excreción proteica en orina de 24 horas y el filtrado glomerular estimado para predecir el riesgo de progresión a diálisis a los 2 años y el riesgo anual. 49. Otros biomarcadores, como el factor de von Willebrand Se ha demostrado que el dímero D,(50 )y el factor de diferenciación de crecimiento (15,52) predicen los resultados y la supervivencia, pero aún no se han incorporado en los sistemas de estadificación.
Gestión
Se han observado aumentos sustanciales en las tasas de supervivencia entre pacientes con amiloidosis AL. Un estudio longitudinal de historia natural que abarcó 40 años reveló una mejora constante en la supervivencia a lo largo del tiempo, con una supervivencia general a 5 años que aumentó del 15 % a mediados de la década de 1980 al 48 % a mediados de la década de 2010(53).
El tratamiento de la amiloidosis AL generalmente implica un enfoque multidisciplinario y debe ser brindado por un equipo médico con experiencia en el tratamiento de esta rara afección, ya que el enfoque puede variar según el alcance y la gravedad de la afectación del órgano(54).
Los tres principios del tratamiento son reducir de forma rápida y sostenible la producción de proteína monoclonal amiloidogénica causante; individualizar la terapia según la afectación de órganos, los efectos tóxicos previstos y la extensión de la enfermedad; y brindar atención de apoyo específica de órganos para minimizar las complicaciones relacionadas con el tratamiento, reducir el riesgo de muerte y maximizar la calidad de vida.
Terapia de apoyo
La atención de apoyo, que requiere la colaboración entre especialistas , tiene como objetivo aliviar los síntomas y preservar la función de los órganos(55).
El tratamiento para la miocardiopatía amiloide incluye restricción de sodio, uso cuidadoso de diuréticos y posible uso de inhibidores de la enzima convertidora de angiotensina (ECA) para la reduction de postcarga. La digoxina generalmente no es beneficiosa excepto en ciertos casos de fibrilación auricular.56.
Se necesita precaución con la anticoagulación debido al riesgo de hemorragia. Por lo general, se evitan los bloqueadores de los canales de calcio. El síncope recurrente puede requerir un marcapaso, mientras que las arritmias ventriculares se tratan con amiodarona o desfibriladores implantables en algunos casos.
La hipotensión ortostática se puede controlar con varias medidas. La fludrocortisona a menudo no es una buena opción debido a la retención de líquidos asociada.
El tratamiento de apoyo para la enfermedad renal asociada a amiloide incluye restricción de sal, diuréticos y tratamiento de la hiperlipidemia. Los inhibidores de la ECA o los bloqueadores de los receptores de angiotensina pueden ayudar con la proteinuria si no están contraindicados por la hipotensión.
La hemodiálisis y la diálisis peritoneal son opciones para la enfermedad renal terminal. Se está explorando el papel de los inhibidores de la proteína transportadora de sodio-glucosa 2 (SGLT2) para la afectación renal y cardíaca. La diarrea es un problema incapacitante para los pacientes con afectación del sistema nervioso autónomo y puede controlarse con ciertos medicamentos.
Una alimentación oral o intravenosa adecuada es esencial para los pacientes desnutridos. El dolor neuropático se puede tratar con gabapentina, duloxetina o pregabalina.
Se pueden utilizar agentes analgésicos no nefrotóxicos como terapia adyuvante. Las complicaciones hemorrágicas son comunes y pueden justificar diversas intervenciones, incluida la esplenectomía por deficiencia adquirida del factor X, así como reemplazos de factores de coagulación.
La deficiencia de hierro debida a la pérdida crónica de sangre en pacientes con diátesis hemorrágica o afectación gastrointestinal debe controlarse y corregirse con infusiones de hierro.
Evaluación de la respuesta al tratamiento
Los criterios para las respuestas hematológicas y orgánicas en pacientes con amiloidosis AL están unificados y formalizados . Un grupo internacional estableció y validó criterios de respuesta hematológica y de órganos.57.
Los criterios para la respuesta de órganos, que anteriormente eran binarios, se han graduado y pueden predecir la supervivencia y los resultados clínicos a largo plazo(58,59).
Se han sugerido mejoras en los criterios de respuesta hematológica con un valor de iFLC (cadena ligera libre involucrada) de menos de 20 mg por litro y un valor de dFLC de menos de 10 mg por litro predicen una supervivencia más larga(60,61).
Los datos emergentes indican la importancia de una enfermedad residual mensurable, que puede ser responsable de disfunción orgánica residual a pesar de una respuesta hematológica de alta calidad(62–64).
Se ha descubierto que una respuesta hematológica temprana y profunda conduce a una supervivencia significativamente prolongada y, por lo tanto, la respuesta hematológica debe medirse cada mes durante el tratamiento.65.
Puede ser evidente una mejor función de los órganos sólo de 6 a 12 meses después del tratamiento, aunque también pueden ocurrir respuestas tardías, que ocurren hasta 24 meses después del tratamiento(59,66).
El tiempo desde el inicio del tratamiento dirigido contra la discrasia de células plasmáticas hasta la mejor respuesta orgánica puede variar: 24 meses para una respuesta cardíaca, 29 meses para una respuesta renal y 35 meses para una respuesta hepática.59.
Tratamiento de la amiloidosis AL recién diagnosticada
El melfalán intravenoso en dosis altas y el autotrasplante de células madre de sangre periférica (SCT) se han utilizado como tratamiento desde mediados de la década de 1990 para pacientes seleccionados con amiloidosis AL.
Numerosos estudios unicéntricos y multicéntricos de SCT han demostrado su eficacia en la amiloidosis AL. El SCT conduce a una respuesta hematológica completa en el 40% de los pacientes, y la duración media de una respuesta completa es de 12,3 años.67.
Con una mediana de seguimiento de 8 años, la mediana de la supervivencia libre de eventos y la supervivencia general se prolongan, a 3,3 y 7,6 años, respectivamente.67.
Los pacientes con una respuesta hematológica completa tuvieron una mediana de supervivencia general de 15 años, y el 30% de estos pacientes sobrevivió durante más de 20 años.67. Sin embargo, sólo del 10 al 20% de los pacientes con amiloidosis AL recién diagnosticada son elegibles para SCT debido a factores como un estado funcional deficiente, disfunción orgánica avanzada y enfermedad multiorgánica. El panorama terapéutico en expansión es otra razón del papel limitado de la SCT.
El Grupo de Trabajo de la Sociedad Internacional de Amiloidosis, que comprende un esfuerzo de colaboración con representación de seis países, ha publicado directrices para el SCT en la amiloidosis AL. Estas directrices cubren los criterios de elegibilidad, las indicaciones para la terapia de inducción, la movilización y recolección de células madre, la dosificación de melfalán adaptada al riesgo y los cuidados de apoyo después del TCM.68.
Los pacientes que no son elegibles para SCT (aproximadamente el 80%) reciben tratamiento con la combinación de ciclofosfamida, bortezomib y dexametasona (CyBorD) más daratumumab, que es la terapia de primera línea preferida según el ensayo ANDROMEDA (Un estudio para evaluar la Eficacia y seguridad de daratumumab en combinación con CyBorD en comparación con CyBorD solo en amiloidosis AL sistémica recién diagnosticada).
CyBorD solo o bortezomib-melfalán-dexametasona se utiliza cuando el acceso a daratumumab es limitado. Daratumumab-CyBorD produce altos porcentajes de respuesta hematológica: el 78 % de los pacientes tiene una respuesta parcial muy buena o mejor y aproximadamente el 50 al 55 % tiene una respuesta orgánica 18 meses después del tratamiento.69.
Se deben considerar ciertas características del paciente al elegir un régimen. Por ejemplo, el tratamiento con la combinación de bortezomib, melfalán y dexametasona(70) puede superar los efectos tanto de la ganancia 1q21 (que confiere un peor resultado con melfalán oral y posiblemente daratumumab)(71) como de t(11;14) (que confiere un peor resultado con bortezomib)(13). Los pacientes con enfermedad de alto riesgo representan aproximadamente el 20% de todos los pacientes con amiloidosis AL y representan un desafío debido a una enfermedad cardíaca avanzada (estadio IIIb) o insuficiencia cardíaca grave (clase III o IV de la New York Heart Association). .72.
Tratamiento de la recaída y progresión después de la terapia inicial
No se ha establecido un consenso sobre los criterios para comenzar la terapia de segunda línea en pacientes con enfermedad progresiva después de la terapia inicial(73,74). Los pacientes con enfermedad recidivante pueden ser tratados repitiendo la terapia de primera línea si la respuesta duró más de un año, aunque estos pacientes tienen un tiempo más corto para recaer sin una reducción en la supervivencia general que los pacientes que son tratados con una terapia diferente para la enfermedad recidivante.
Las posibles opciones disponibles para el tratamiento de la amiloidosis AL sistémica recidivante incluyen inhibidores del proteasoma,(75,76), anticuerpos monoclonales anti-CD-38(77,78), agentes inmunomoduladores(79), venetoclax para pacientes con t(11;14)(80) ,bendamustina(81), alta -dosis de melfalán con SCT autólogo(82,83 ), anticuerpos biespecíficos(84,85), e incluso terapia de células T con receptor de antígeno quimérico(86).
Aunque no es posible ser prescriptivo con respecto a la secuenciación de las terapias, las dos consideraciones rectoras son la profundidad y la duración. de la respuesta inicial y la elección de una clase de agentes no utilizados previamente. También se deben considerar las limitaciones impuestas por el nivel reducido de aptitud física o fragilidad del paciente y el daño a los órganos terminales. Se fomenta la inscripción en ensayos clínicos.
Anticuerpos monoclonales antifibrillas
La quimioterapia se dirige a la discrasia de las células plasmáticas y la producción de proteína precursora amiloidogénica, pero no reabsorbe ni degrada directamente los depósitos de amiloide en los tejidos.
Aunque los marcadores de disfunción orgánica mejoran con la supresión del precursor amiloide, actualmente se están investigando dos anticuerpos, birtamimab y anselamimab, como agentes antifibrillas.
El dezamizumab (un anticuerpo anti-componente P amiloide en suero), que alguna vez estuvo bajo consideración, ya no se investiga. Los anticuerpos antifibrillas tienen el potencial de eliminar las fibrillas de amiloide de los órganos activando las células inmunes para la degradación química y enzimática e induciendo la fagocitosis dependiente de anticuerpos.87.
Birtamimab (NEOD0001) es un anticuerpo monoclonal completamente humanizado que se dirige a un epítopo críptico en la proteína amiloide A sérica que se revela cuando está mal plegado.
Este agente tiene una reacción cruzada con las fibrillas de amiloide de cadena ligera de inmunoglobulina y, según se informa, activa la degradación mediada por macrófagos y la eliminación de las fibrillas de cadena ligera(88). Un análisis post hoc no preespecificado de los datos del estudio VITAL (A Fase 3, aleatorizado, multicéntrico, doble ciego,
Estudio de eficacia y seguridad de 2 brazos, controlado con placebo, de NEOD001 Plus Standard of Care versus Placebo Plus Standard of Care en sujetos con amiloidosis AL, que finalizó anticipadamente sobre la base de un análisis de inutilidad provisional demostró un beneficio de supervivencia con birtamimab en pacientes con amiloidosis AL cardíaca avanzada en estadio IV.
El estudio internacional AFFIRM-AL (A Study to Evaluate the Efficacy and Safety of Birtamimab in Mayo Stage IV Patients with AL Amyloidosis; NCT04973137), un ensayo de fase 3 aleatorizado, doble ciego y controlado con placebo diseñado para confirmar este hallazgo, está en curso. .
Anselamimab (CAEL-101) es un anticuerpo monoclonal quimérico que se dirige a un epítopo críptico de las cadenas ligeras de inmunoglobulinas que queda expuesto cuando las cadenas ligeras están mal plegadas.
Se une a cadenas ligeras de inmunoglobulinas libres mal plegadas, así como a fibrillas de amiloide depositadas en los órganos.
Se plantea la hipótesis de que CAEL-101 opsoniza las fibrillas de amiloide y las cadenas ligeras mal plegadas, atrayendo y activando así macrófagos que degradan el complejo mediante fagocitosis, proteólisis enzimática y química, o ambas(89).
Dos ensayos de fase 3, aleatorizados, doble ciego, están diseñados para evaluar la eficacia y seguridad de la coadministración de CAEL-101 con el tratamiento estándar para la discrasia de células plasmáticas en pacientes con amiloidosis AL y miocardiopatía grave en estadios IIIa y IIIb (Un estudio para evaluar la eficacia y seguridad de CAEL-101 en Pacientes con amiloidosis AL en estadio IIIa de Mayo [NCT04512235] y un estudio para evaluar la eficacia y seguridad de CAEL-101 en pacientes con amiloidosis AL en estadio IIIb de Mayo [NCT04504825]) .
Direcciones futuras
El progreso que se ha logrado en el tratamiento de la amiloidosis AL no tiene precedentes. Los esfuerzos futuros deben centrarse en aumentar el diagnóstico temprano a través de la educación profesional, definir el estándar de atención para pacientes con enfermedad cardíaca avanzada, definir la progresión hematológica o de órganos que justifica un tratamiento adicional, evaluar nuevas técnicas para evaluar la respuesta hematológica de manera más estricta, investigar nuevas técnicas dirigidas por clones y el desarrollo de tratamientos dirigidos a las cadenas ligeras mal plegadas(90) y a los depósitos de fibrillas de amiloide.
Conclusiones
Hay tratamientos prometedores disponibles para pacientes con amiloidosis AL. El diagnóstico oportuno y la derivación adecuada tienen el potencial de mejorar la supervivencia y los resultados de estos pacientes.
La inclusión de la amiloidosis AL en el diagnóstico diferencial de pacientes evaluados por diversos síndromes, en particular pacientes con proteinuria en rango nefrótico, miocardiopatía no isquémica inexplicable, neuropatía periférica, hepatomegalia inexplicable o mieloma múltiple atípico, debería mejorar la eficiencia del diagnóstico. A pesar de las mejoras en el diagnóstico y tratamiento de la amiloidosis AL, se necesitan esfuerzos continuos de investigación básica y clínica para iluminar el futuro de los pacientes con este trastorno.
Referencias Bibliográficas
1. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers 2018;4:38-38.
2. Brenner DA, Jain M, Pimentel DR, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 2004;94:1008-1010.
3. Buxbaum JN, Dispenzieri A, Eisenberg DS, et al. Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2022;29:213-219.
4. Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006;108:2520-2530.
5. Muchtar E, Dispenzieri A, Magen H, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med 2021;289:268-292.
6. Koike H, Katsuno M. Transthyretin amyloidosis: update on the clinical spectrum, pathogenesis, and disease-modifying therapies. Neurol Ther 2020;9:317-333.
7. Morgan GJ, Wall JS. The process of amyloid formation due to monoclonal immunoglobulins. Hematol Oncol Clin North Am 2020;34:1041-1054.
8. Diomede L, Rognoni P, Lavatelli F, et al. A Caenorhabditis elegans-based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood 2014;123:3543-3552.
9. Mishra S, Guan J, Plovie E, et al. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am J Physiol Heart Circ Physiol 2013;305:H95-H103.
10. Hipp MS, Park S-H, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 2014;24:506-514.
11. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997;337:898-909.
12. Nasr SH, Said SM, Valeri AM, et al. The diagnosis and characteristics of renal heavy-chain and heavy/light-chain amyloidosis and their comparison with renal light-chain amyloidosis. Kidney Int 2013;83:463-470.
13. Bochtler T, Hegenbart U, Kunz C, et al. Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J Clin Oncol 2015;33:1371-1378.
14. Bochtler T, Hegenbart U, Heiss C, et al. Hyperdiploidy is less frequent in AL amyloidosis compared with monoclonal gammopathy of undetermined significance and inversely associated with translocation t(11;14). Blood 2011;117:3809-3815.
15. Morgan GJ, Kelly JW. The kinetic stability of a full-length antibody light chain dimer determines whether endoproteolysis can release amyloidogenic variable domains. J Mol Biol 2016;428:4280-4297.
16. Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med 2018;378:241-249.
17. Madan S, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc 2010;85:232-238.
18. Weiss BM, Hebreo J, Cordaro DV, et al. Increased serum free light chains precede the presentation of immunoglobulin light chain amyloidosis. J Clin Oncol 2014;32:2699-2704.
19. Munshi NC. Association of Agent Orange with plasma cell disorder: further evidence. JAMA Oncol 2015;1:1035-1036.
20. Kumar S, Murray D, Dasari S, et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: a potential path to earlier AL diagnosis for a subset of patients. Leukemia 2019;33:254-257.
21. Kourelis T, Murray DL, Dasari S, et al. MASS-FIX may allow identification of patients at risk for light chain amyloidosis before the onset of symptoms. Am J Hematol 2018;93(11):E368-E370.
22. Kyle RA, Larson DR, Kurtin PJ, et al. Incidence of AL amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin Proc 2019;94:465-471.
23. Kumar N, Zhang NJ, Cherepanov D, Romanus D, Hughes M, Faller DV. Global epidemiology of amyloid light-chain amyloidosis. Orphanet J Rare Dis 2022;17:278-278.
24. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv 2018;2:1046-1053.
25. McCausland KL, White MK, Guthrie SD, et al. Light chain (AL) amyloidosis: the journey to diagnosis. Patient 2018;11:207-216.
26. Gupta N, Kaur H, Wajid S. Renal amyloidosis: an update on diagnosis and pathogenesis. Protoplasma 2020;257:1259-1276.
27. Merlini G, Lousada I, Ando Y, et al. Rationale, application and clinical qualification for NT-proBNP as a surrogate end point in pivotal clinical trials in patients with AL amyloidosis. Leukemia 2016;30:1979-1986.
28. Sayed RH, Rogers D, Khan F, et al. A study of implanted cardiac rhythm recorders in advanced cardiac AL amyloidosis. Eur Heart J 2015;36:1098-1105.
29. Comenzo RL, Zhang Y, Martinez C, Osman K, Herrera GA. The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001;98:714-720.
30. Kourelis TV, Dasari S, Theis JD, et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017;129:299-306.
31. Muchtar E, Dispenzieri A, Lacy MQ, et al. Overuse of organ biopsies in immunoglobulin light chain amyloidosis (AL): the consequence of failure of early recognition. Ann Med 2017;49:545-551.
32. Sanchorawala V, Blanchard E, Seldin DC, O’Hara C, Skinner M, Wright DG. AL amyloidosis associated with B-cell lymphoproliferative disorders: frequency and treatment outcomes. Am J Hematol 2006;81:692-695.
33. Schönland SO, Hegenbart U, Bochtler T, et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 2012;119:488-493.
34. Fernández de Larrea C, Verga L, Morbini P, et al. A practical approach to the diagnosis of systemic amyloidoses. Blood 2015;125:2239-2244.
35. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR III, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 2009;114:4957-4959.
36. Dasari S, Theis JD, Vrana JA, et al. Amyloid Typing by mass spectrometry in clinical practice: a comprehensive review of 16,175 samples. Mayo Clin Proc 2020;95:1852-1864.
37. Phull P, Sanchorawala V, Connors LH, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid 2018;25:62-67.
38. Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis. Evidence base and standardized methods of imaging. Circ Cardiovasc Imaging 2021;14(7):e000029-e000029.
39. Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis. 2. Diagnostic criteria and appropriate utilization. Circ Cardiovasc Imaging 2021;14(7):e000030-e000030.
40. Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 2004;22:3751-3757.
41. Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood 2013;121:3420-3427.
42. Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012;30:989-995.
43. Khwaja J, Ravichandran S, Bomsztyk J, et al. Limited utility of Mayo 2012 cardiac staging system for risk stratification of patients with advanced cardiac AL amyloidosis — analysis of a uniformly treated cohort of 1,275 patients. Haematologica 2024;109:1598-1602.
44. Green D. A new staging system for light chain amyloidosis. NEJM J Watch. 2018 ().
45. Lilleness B, Doros G, Ruberg FL, Sanchorawala V. Establishment of brain natriuretic peptide-based criteria for evaluating cardiac response to treatment in light chain (AL) amyloidosis. Br J Haematol 2020;188:424-427.
46. Dittrich T, Bochtler T, Kimmich C, et al. AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have an excellent prognosis. Blood 2017;130:632-642.
47. Nguyen VP, Rosenberg A, Mendelson LM, Comenzo RL, Varga C, Sanchorawala V. Outcomes of patients with AL amyloidosis and low serum free light chain levels at diagnosis. Amyloid 2018;25:156-159.
48. Milani P, Basset M, Russo F, Foli A, Merlini G, Palladini G. Patients with light-chain amyloidosis and low free light-chain burden have distinct clinical features and outcome. Blood 2017;130:625-631.
49. Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014;124:2325-2332.
50. Kastritis E, Papassotiriou I, Terpos E, et al. Clinical and prognostic significance of serum levels of von Willebrand factor and ADAMTS-13 antigens in AL amyloidosis. Blood 2016;128:405-409.
51. Pudusseri A, Sanchorawala V, Sloan JM, et al. Prevalence and prognostic value of D-dimer elevation in patients with AL amyloidosis. Am J Hematol 2019;94:1098-1103.
52. Kastritis E, Papassotiriou I, Merlini G, et al. Growth differentiation factor-15 is a new biomarker for survival and renal outcomes in light chain amyloidosis. Blood 2018;131:1568-1575.
53. Staron A, Zheng L, Doros G, et al. Marked progress in AL amyloidosis survival: a 40-year longitudinal natural history study. Blood Cancer J 2021;11:139-139.
54. Kittleson MM, Ruberg FL, Ambardekar AV, et al. 2023 ACC expert consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol 2023;81:1076-1126.
55. Cibeira MT, Ortiz-Pérez JT, Quintana LF, Fernádez de Larrea C, Tovar N, Bladé J. Supportive care in AL Amyloidosis. Acta Haematol 2020;143:335-342.
56. Muchtar E, Gertz MA, Kumar SK, et al. Digoxin use in systemic light-chain (AL) amyloidosis: contra-indicated or cautious use? Amyloid 2018;25:86-92.
57. Palladini G, Schönland SO, Sanchorawala V, et al. Clarification on the definition of complete haematologic response in light-chain (AL) amyloidosis. Amyloid 2021;28:1-2.
58. Muchtar E, Dispenzieri A, Wisniowski B, et al. Graded cardiac response criteria for patients with systemic light chain amyloidosis. J Clin Oncol 2023;41:1393-1403.
59. Muchtar E, Dispenzieri A, Leung N, et al. Depth of organ response in AL amyloidosis is associated with improved survival: grading the organ response criteria. Leukemia 2018;32:2240-2249.
60. Manwani R, Cohen O, Sharpley F, et al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood 2019;134:2271-2280.
61. Sarosiek S, Zheng L, Sloan JM, Quillen K, Brauneis D, Sanchorawala V. Comparing measures of hematologic response after high-dose melphalan and stem cell transplantation in AL amyloidosis. Blood Cancer J 2020;10:88-88.
62. Staron A, Burks EJ, Lee JC, Sarosiek S, Sloan JM, Sanchorawala V. Assessment of minimal residual disease using multiparametric flow cytometry in patients with AL amyloidosis. Blood Adv 2020;4:880-884.
63. Palladini G, Paiva B, Wechalekar A, et al. Minimal residual disease negativity by next-generation flow cytometry is associated with improved organ response in AL amyloidosis. Blood Cancer J 2021;11:34-34.
64. Dispenzieri A, Arendt B, Dasari S, et al. Blood mass spectrometry detects residual disease better than standard techniques in light-chain amyloidosis. Blood Cancer J 2020;10:20-20.
65. Ravichandran S, Cohen OC, Law S, et al. Impact of early response on outcomes in AL amyloidosis following treatment with frontline Bortezomib. Blood Cancer J 2021;11:118-118.
66. Szalat R, Sarosiek S, Havasi A, Brauneis D, Sloan JM, Sanchorawala V. Organ responses after highdose melphalan and stemcell transplantation in AL amyloidosis. Leukemia 2021;35:916-919.
67. Gustine JN, Staron A, Szalat RE, et al. Predictors of hematologic response and survival with stem cell transplantation in AL amyloidosis: a 25-year longitudinal study. Am J Hematol 2022;97:1189-1199.
68. Sanchorawala V, Boccadoro M, Gertz M, et al. Guidelines for high dose chemotherapy and stem cell transplantation for systemic AL amyloidosis: EHA-ISA working group guidelines. Amyloid 2022;29:1-7.
69. Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med 2021;385:46-58.
70. Kastritis E, Leleu X, Arnulf B, et al. Bortezomib, melphalan, and dexamethasone for light-chain amyloidosis. J Clin Oncol 2020;38:3252-3260.
71. Szalat RE, Gustine J, Sloan JM, Edwards CV, Sanchorawala V. Predictive factors of outcomes in patients with AL amyloidosis treated with daratumumab. Am J Hematol 2022;97:79-89.
72. Gustine JN, Staron A, Mendelson L, et al. Predictors of treatment response and survival outcomes in patients with advanced cardiac AL amyloidosis. Blood Adv 2023;7:6080-6091.
73. Sarosiek S, Sanchorawala V. Treatment options for relapsed/refractory systemic light-chain (AL) amyloidosis: current perspectives. J Blood Med 2019;10:373-380.
74. Tandon N, Sidana S, Gertz MA, et al. Treatment patterns and outcome following initial relapse or refractory disease in patients with systemic light chain amyloidosis. Am J Hematol 2017;92:549-554.
75. Dubrey SW, Reece DE, Sanchorawala V, et al. Bortezomib in a phase 1 trial for patients with relapsed AL amyloidosis: cardiac responses and overall effects. QJM 2011;104:957-970.
76. Dispenzieri A, Kastritis E, Wechalekar AD, et al. A randomized phase 3 study of ixazomib-dexamethasone versus physician’s choice in relapsed or refractory AL amyloidosis. Leukemia 2022;36:225-235.
77. Roussel M, Merlini G, Chevret S, et al. A prospective phase 2 trial of daratumumab in patients with previously treated systemic light-chain amyloidosis. Blood 2020;135:1531-1540.
78. Sanchorawala V, Sarosiek S, Schulman A, et al. Safety, tolerability, and response rates of daratumumab in relapsed AL amyloidosis: results of a phase 2 study. Blood 2020;135:1541-1547.
79. Sanchorawala V, Doros G, Shelton AC. Long term outcome of patients treated on clinical trials of immunomodulatory agents for the treatment of Immunoglobulin light chain (AL) amyloidosis: a pooled analysis. Am J Hematol 2019;94(7):E194-E196.
80. Premkumar VJ, Lentzsch S, Pan S, et al. Venetoclax induces deep hematologic remissions in t(11;14) relapsed/refractory AL amyloidosis. Blood Cancer J 2021;11:10-10.
81. Lentzsch S, Lagos GG, Comenzo RL, et al. Bendamustine with dexamethasone in relapsed/refractory systemic light-chain amyloidosis: results of a phase II study. J Clin Oncol 2020;38:1455-1462.
82. Quillen K, Seldin DC, Finn KT, Sanchorawala V. A second course of high-dose melphalan and auto-SCT for the treatment of relapsed AL amyloidosis. Bone Marrow Transplant 2011;46:976-980.
83. Abdallah N, Sidana S, Dispenzieri A, et al. Outcomes with early vs. deferred stem cell transplantation in light chain amyloidosis. Bone Marrow Transplant 2020;55:1297-1304.
84. Chakraborty R, Bhutani D, Maurer MS, Mohan M, Lentzsch S, D’Souza A. Safety and efficacy of teclistamab in systemic immunoglobulin light chain amyloidosis. Blood Cancer J 2023;13:172-172.
85. Forgeard N, Elessa D, Carpinteiro A, et al. Teclistamab in relapsed or refractory AL amyloidosis: a multinational retrospective case series. Blood 2024;143:734-737.
86. Kfir-Erenfeld S, Asherie N, Grisariu S, et al. Feasibility of a novel academic BCMA-CART (HBI0101) for the treatment of relapsed and refractory AL amyloidosis. Clin Cancer Res 2022;28:5156-5166.
87. Nuvolone M, Nevone A, Merlini G. Targeting amyloid fibrils by passive immunotherapy in systemic amyloidosis. BioDrugs 2022;36:591-608.
88. Gertz MA, Cohen AD, Comenzo RL, et al. Birtamimab plus standard of care in light-chain amyloidosis: the phase 3 randomized placebo-controlled VITAL trial. Blood 2023;142:1208-1218.
89. Edwards CV, Rao N, Bhutani D, et al. Phase 1a/b study of monoclonal antibody CAEL-101 (11-1F4) in patients with AL amyloidosis. Blood 2021;138:2632-2641.
90. Morgan GJ, Yan NL, Mortenson DE, et al. Stabilization of amyloidogenic immunoglobulin light chains by small molecules. Proc Natl Acad Sci U S A 2019;116:8360-8369.