Ronald Palacios Castrillo, M.D.,PhD.
PREFACIO
No se hace mucho enfásis en que el cáncer del tipo y estadío clínico que sea, es en realidad una enfermedad sistémica y como tal ,se debe abordar y tratar.
Hace 7 meses o más decidí estudiar y revisar (no de forma exclusiva ni cotidiana) la extensa bibliografía sobre este tema (259 referencias y 6 figuras) y de plasmar las secciones de esta revisión que está escrita de una manera muy accesible tanto para los profesionales en salud, como también para los que no, pero que son interesados y curiosos de las ciencias biológicas.
Resumen
=> Recibir por Whatsapp las noticias destacadas
Los últimos 50 años han sido testigos de avances extraordinarios en la comprensión de los mecanismos de carcinogénesis, sintetizados como las características distintivas del cáncer.
A pesar de este marco lógico, nuestra comprensión de las bases moleculares de las manifestaciones sistémicas y las causas subyacentes de la muerte relacionada con el cáncer sigue siendo incompleta.
De cara al futuro, dilucidar cómo interactúan los tumores con órganos distantes y cómo los parámetros ambientales y fisiológicos multifacéticos inciden en los tumores y sus huéspedes será crucial para avanzar en la prevención y el tratamiento más eficaz de los cánceres humanos.
En esta perspectiva, analizamos las complejidades del cáncer como enfermedad sistémica, incluida la iniciación y promoción del tumor, los microambientes y macroambientes inmunes del tumor, el envejecimiento, el metabolismo y la obesidad, la caquexia del cáncer, los ritmos circadianos, las interacciones del sistema nervioso, la trombosis relacionada con el tumor, y el microbioma.
Los sistemas modelo que incorporen la variación genética humana serán esenciales para descifrar la base mecanicista de estos fenómenos y desentrañar las interacciones entre genes y medio ambiente, proporcionando una síntesis moderna de oncología molecular preparada para prevenir cánceres y mejorar la calidad de vida de los pacientes y los resultados del cáncer.
Introducción
Mirando hacia atrás 50 años, es evidente que el panorama de la investigación del cáncer ha cambiado dramáticamente.
Durante este tiempo, la biología celular reduccionista que utiliza sistemas modelo elegantes y simples ha formado el pilar del descubrimiento científico, produciendo conocimientos extraordinarios sobre la regulación del ciclo celular, la apoptosis, la motilidad celular, la invasión y la desregulación inmune, lo que ha llevado a un progreso significativo en el diagnóstico. y tratamiento del cáncer; Las tasas de supervivencia a 5 años han aumentado del 35 % en la década de 1950 al 69,7 % en 2017 (SEER Cancer Statistics Review 1975-2018). A pesar de este progreso, el cáncer sigue siendo una de las principales causas de muerte en todo el mundo.
Estamos en medio de un renacimiento en el que el conocimiento sobre la biología y la genética del cáncer se está disparando, con más de 1000 genes identificados que están alterados en los tumores, ya sea genéticamente por mutaciones recurrentes o epigenéticamente, lo que resulta en cambios en su regulación y expresión.1.
Este hito ha facilitado una delimitación cada vez más precisa de los circuitos moleculares, las constituciones celulares y la dinámica poblacional heterogénea de las células cancerosas mutantes, así como los mecanismos de progresión tumoral y diseminación metastásica en conjunto con células aparentemente normales, pero funcionalmente corruptas, de orígenes distintivos.
Este conocimiento, que tiende a centrarse en la propia célula cancerosa y su microambiente local, ha permitido nuevas generaciones de fármacos terapéuticos y regímenes de tratamiento guiados por mecanismos que han beneficiado a algunos pacientes con determinadas formas de cáncer.
Es frustrante que pocas de estas nuevas estrategias terapéuticas innovadoras sean ampliamente beneficiosas en todo el espectro de cánceres humanos y, con muchas vías para que los tumores desarrollen resistencia, aún menos prolongan significativamente la supervivencia general.
La arqueología multidimensional del cáncer, combinada con el avance de las tecnologías, crea un ámbito vertiginosamente vasto para los “grandes datos” hasta el nivel unicelular.
Estas tecnologías tienen como objetivo caracterizar los cánceres con cada vez más detalle, arrojando luz sobre el desarrollo neodarwiniano y la progresión de los cánceres de diferentes células de origen que enfrentan barreras específicas de tejido que requieren adaptaciones distintivas reflejadas en su tumorigénesis de múltiples pasos. La apreciación de esta aleccionadora diversidad y complejidad ha ido creciendo durante décadas, y se perfila como un desafío potencialmente insuperable para la “guerra contra el cáncer”.
Hace 24 años, se publicó un intento de conciliar esta enorme diversidad con el creciente conocimiento sobre los mecanismos del cáncer.2.
Las características del cáncer postulaban que prácticamente todos los tumores adquieren un conjunto común de capacidades funcionales cualitativamente distintas que colectivamente permiten que las células cancerosas proliferen expansivamente mientras orquestan la formación de tumores que crecen y a menudo se diseminan. El corolario del concepto fue que un par común de características fenotípicas (inestabilidad genómica y mutación, junto con inflamación) facilitan su adquisición.
Por lo tanto, se argumentó que la inmensa complejidad de la patogénesis del cáncer podría resumirse en diferentes soluciones al mismo desafío, es decir, adquirir el mismo conjunto de capacidades distintivas durante la tumorigénesis y la progresión maligna. Este concepto simple, refinado en los años siguientes(3,4,5), ha resonado en la medicina del cáncer hasta el día de hoy, lo que indica que tiene cierta utilidad como principio organizador conceptual.
Sin embargo, esta síntesis moderna de la biología del cáncer no considera plenamente las interacciones más amplias de un tumor en evolución con órganos distantes del huésped, ni el impacto de la fisiopatología del huésped, la diversidad genética de la línea germinal y las exposiciones ambientales en el inicio y la evolución del cáncer.
La tranquilizadora simplicidad de las características es claramente insuficiente para comprender completamente las múltiples manifestaciones de los mecanismos de la enfermedad.
Como tal, existe una clara necesidad de desarrollar nuevas estrategias terapéuticas que mejoren tanto la calidad como la duración de la vida de los pacientes que padecen cáncer, abordando algunas de las afecciones más peligrosas para la vida, como la caquexia por cáncer, la trombosis y los síndromes paraneoplásicos y, lo que es más importante, oportunidades para intervenir en las primeras etapas del inicio del cáncer en nuevas estrategias de prevención.
Un repertorio en expansión de herramientas de investigación (multiómicas) y sistemas modelo refinados están ahora preparados para abordar el cáncer como una enfermedad sistémica resultante de la compleja interacción entre la diversidad del genoma del huésped, los eventos fortuitos y un legado del comportamiento humano que resulta en complejos exposiciones ambientales.
El envejecimiento es el factor pronóstico número uno para la mayoría de los tumores, dado que más del 90% de los pacientes diagnosticados con cáncer tienen más de 50 años.
Además de este factor importante que contribuye a la complejidad del cáncer, y de hecho una ruta para estrategias preventivas viables, está el medio ambiente en el que vivimos, que según estudios epidemiológicos se estima que influye en el desarrollo de hasta el 80% de los cánceres humanos.6.
El término “medio ambiente” abarca una amplia gama de factores exógenos, incluidos los contaminantes industriales en el aire o en nuestra dieta, así como exposiciones ocupacionales o médicas a sustancias tóxicas o ciertos tipos de radiación e infecciones virales y bacterianas patógenas.
También incluye lo que se conoce como “factores de estilo de vida”: dieta, consumo de alcohol, tabaquismo, exposición al sol y comportamiento sedentario, todos los cuales influyen en el riesgo de cáncer.
Sorprendentemente, cada vez está más claro que estos agentes ambientales dispares aumentan el riesgo de cáncer al afectar las mismas características distintivas del cáncer descritas anteriormente: mutaciones genómicas, metabolismo alterado, inestabilidad cromosómica e inflamación.
En esta perspectiva, culminación de cuatro Simposios Marshall patrocinados por Cancer-Research-UK, contemplamos, como se ilustra en la Figura 1, la nueva frontera que complementa las características distintivas del cáncer al abarcar la complejidad de la patogénesis del cáncer humano, utilizando avances tecnológicos para desentrañar la interacción diversa entre un tumor en evolución dentro de un entorno que envejece y sistemas de órganos distantes en poblaciones genéticamente diversas que han tenido comportamientos distintos y han estado expuestos a exposiciones ambientales heterogéneas.
Es importante destacar que sostenemos que durante los próximos 50 años necesitaremos reconocer las limitaciones de los modelos animales endogámicos en ambientes altamente controlados para recapitular completamente tal complejidad.
Abordar este desafío requerirá estudios bien concebidos en sujetos humanos como vehículo para avanzar en el descubrimiento, interconectados con modelos ex vivo y animales cada vez más sofisticados de las complejidades del cáncer, con el fin de avanzar en la intervención terapéutica y la interceptación temprana para prevenir, diagnosticar y tratar el cáncer de manera más efectiva. y sus manifestaciones sistémicas.
Aunque las características distintivas del cáncer han proporcionado una justificación conceptual general para las innumerables manifestaciones que abarcan el cáncer como enfermedad, debajo de esta simplicidad se esconde una vertiginosa diversidad de efectos mecanicistas y fenotipos, tanto dentro de los tumores como a nivel de todo el sistema en el individuo afectado.
Por lo tanto, sobre el horizonte hay nubes de complejidad que, argumentamos desde esta perspectiva, son importantes y no se comprenden completamente.
Debajo del horizonte se encuentran efectores mecanicistas (los componentes básicos del cáncer) que gobiernan el inicio y la progresión del cáncer, que tampoco se comprenden completamente. Esclarecer ambas dimensiones del cáncer como enfermedad sistémica será fundamental para lograr innovaciones revolucionarias en la prevención y el tratamiento duradero del cáncer humano.
Cualquier revisión con el objetivo visionario de trazar direcciones futuras para la investigación del cáncer en los próximos 50 años bien podría considerar otros temas destacados que no se destacan en la Figura 1.
Entre ellos destaca el papel del sexo biológico en la susceptibilidad, el desarrollo y las respuestas terapéuticas al cáncer.
Aparte de la importancia de las hormonas sexuales en el desarrollo de los cánceres de mama, ovario y próstata, el sexo biológico se asocia con distintas incidencias de una variedad de otros tipos de cáncer, incluidos los de vejiga, riñón y esófago(7,8), todos los cuales son más altos en hombres.
Cada vez hay más pruebas que respaldan el papel de los efectos de los genes de los cromosomas sexuales(9) y la interferencia entre las hormonas sexuales y otros sistemas implicados en la inflamación y la inmunidad(10,11), efectos que tienen implicaciones claras para el tratamiento del cáncer y las respuestas terapéuticas.
Por ejemplo, comprender por qué el cáncer de pulmón en quienes nunca han fumado es más común en las mujeres requerirá desentrañar el papel del medio ambiente, la genética y el comportamiento humano. Este campo está maduro para un análisis más profundo en las próximas décadas.
Sería negligente discutir la complejidad del cáncer sin mencionar cómo el ritmo cada vez mayor del desarrollo de algoritmos de inteligencia artificial (IA) puede afectar nuestra capacidad para procesar las grandes cantidades de datos que se generan y comprender cómo las propiedades emergentes de las redes interconectadas de genes puede ayudarnos a comprender la progresión del cáncer desde células iniciadas hasta metástasis a nivel unicelular.
En última instancia, anticipamos un momento en el que estos nuevos métodos se aplicarán a múltiples dimensiones de la prevención y el tratamiento del cáncer, desde el análisis de la arquitectura clonal de los tejidos normales hasta los signos esclarecedores de un mayor riesgo y la predicción de los estados de plasticidad de las células madre que conducen a resistencia a los medicamentos y mala supervivencia del paciente.
Incluso cuando se comprendan estas nubes de complejidad y la IA esté preparada para mejorar los resultados para los pacientes, es posible que nunca se mitigue por completo el papel de la “mala suerte” en las mutaciones que inician el cáncer.
Por último, centrarse en la complejidad del cáncer no debería restar valor a los enfoques reduccionistas para comprender las características distintivas del cáncer, particularmente con respecto a la naturaleza dinámica y evolutiva de la enfermedad en el espacio y el tiempo.
Estudios de muestreo longitudinal profundo integrados con programas de autopsia para descifrar la coevolución del tumor dentro de su microambiente durante el curso de la enfermedad y en sitios metastásicos distantes, ejemplificados por estudios como TRACERx renal, pulmonar y mamario, TRACERx EVO de pulmón y el Reino Unido.
El programa nacional de autopsias PEACE, están preparados para ayudar con estos esfuerzos para crear «atlas de tumores dinámicos» en todos los subtipos de cáncer. Sin embargo, consideraciones de espacio no nos permiten considerar estas importantes áreas con la profundidad que merecen.
Aquí, trazamos una hoja de ruta que delinea una selección de temas generales (nubes de complejidad) que se vislumbran en el horizonte de la biología y la medicina del cáncer y que, si se abordan mecánicamente, prometen mejorar los resultados y la calidad de vida de los pacientes.
Tumorigénesis de varios pasos: comprensión de la promoción y progresión del tumor
Desde hace tiempo se sabe que los cánceres se desarrollan a través de vías de tumorigénesis gradual y progresión maligna y, más recientemente, durante la resistencia adaptativa a la terapia.
En parte, estas transiciones y etapas graduales reflejan la adquisición y el refinamiento de capacidades distintivas, habilitadas en particular a través de las características fenotípicas prominentes de la inestabilidad del genoma y la mutación genética, y la inflamación de los tejidos(2,4).
La genética del cáncer nos ha enseñado que las mutaciones en genes reguladores específicos (denominados oncogenes, de los cuales los genes RAS son prototipos) son impulsores esenciales de esta enfermedad.
Junto con las mutaciones de pérdida de función en los genes supresores de tumores, estos eventos genéticos complementarios pueden conducir a la adquisición de múltiples capacidades características del cáncer que se encuentran en la mayoría de los cánceres humanos.
De hecho, este paradigma ha sido fuertemente respaldado por elegantes modelos de cáncer en ratones en los que las modificaciones genéticas somáticas o de la línea germinal pueden combinarse para desencadenar el conjunto completo de fenotipos asociados con los cánceres humanos, en ausencia de factores carcinogénicos ambientales evidentes.
Sin embargo, nuevos conocimientos han revelado que los tejidos aparentemente “normales” de todo el cuerpo albergan una multitud de eventos impulsores oncogénicos que comprenden un mosaico de células mutantes que persisten y pueden expandirse como clones asintomáticos durante el envejecimiento(12,13). Este provocativo resultado nos informa que las mutaciones en Los oncogenes «impulsores» pueden ser necesarios, pero no siempre suficientes, para la tumorigénesis (Figura 2).
Se estima que aproximadamente el 40% de la población desarrollará cáncer a lo largo de su vida y, dado que un ser humano típico tiene 30 billones de células, estas observaciones suscitan la pregunta: ¿por qué el cáncer es tan raro a nivel unicelular?
Las mutaciones son esenciales para el desarrollo de cánceres y pueden surgir como resultado de errores espontáneos en la replicación normal del ADN, durante el envejecimiento de células inactivas o por exposición a mutágenos en el medio ambiente.
La obesidad, los factores dietéticos y la inflamación también pueden contribuir indirectamente a la carga de mutaciones, por ejemplo, a través de la generación de especies reactivas de oxígeno (ROS), pero es poco probable que esto contribuya de manera importante a las cifras generales de mutaciones.
Las mutaciones pueden permanecer latentes en los tejidos normales durante largos períodos, a menos que el tejido se exponga repetidamente a un inductor de inflamación o lesión tisular, lo que provoca la selección de células portadoras de mutaciones específicas, lo que lleva a un crecimiento clonal.
Las mutaciones particulares seleccionadas dependen de muchos factores, incluido el tejido o célula de origen, los antecedentes genéticos del huésped o la naturaleza del factor promotor específico. Se muestran como ejemplos factores promotores exógenos conocidos o sospechados, así como posibles factores promotores endógenos o asociados al estilo de vida.
El concepto histórico de que la carcinogénesis cutánea inducida químicamente, imitando agresiones ambientales naturales, implica “iniciar” y “promover” eventos(14) se ha refinado conceptualmente al darse cuenta de que muchos tejidos normales están repletos de células que contienen mutaciones latentes en oncogenes y genes supresores de tumores(12,13).
Ciertamente, algunos carcinógenos ambientales actúan sinérgicamente como mutágenos e inductores de inflamación (ejemplificados por el humo del tabaco) para iniciar y promover mutacionalmente el cáncer.
Esta sugerencia, hecha por primera vez en la década de 1940, tiene implicaciones importantes para la prevención del cáncer y suposiciones no comprobadas con respecto a la seguridad a largo plazo de los cigarrillos electrónicos(15).
Sin embargo, Riva y sus colegas demostraron recientemente que muchos carcinógenos iniciadores conocidos o sospechados no parecen no causar mutaciones en sí mismas, sino que pueden actuar estimulando otras características (incluidas, entre otras, la inflamación crónica) que despiertan y desencadenan la expansión clonal de células que portan mutaciones oncogénicas latentes(16).
El concepto de que los factores no mutacionales (por ejemplo, la cicatrización de heridas, inflamación y exposición a sustancias químicas en el medio ambiente) puede estimular el crecimiento y la selección de células que contienen tales mutaciones activadoras en oncogenes o mutaciones inactivadoras en genes supresores de tumores, como se ha documentado en modelos de cáncer de ratón,(16,17,18,19,20) en humanos cánceres de pulmón relacionados con la contaminación del aire(21) y en el mesotelioma debido a la exposición al asbesto(22) y está implicado en otros cánceres humanos, incluidos los que surgen en el esófago(23), el páncreas(24) y el colon(25).
Una implicación lógica y pasada por alto de estos importantes conocimientos es que carecemos de ensayos biológicos para evaluar la posible “actividad promotora de tumores” de materia química existente o nueva introducida en el medio ambiente.
Prevemos la necesidad de trazar la totalidad de las vías de múltiples etapas de la tumorigénesis en los tejidos, para aclarar los mecanismos mediante los cuales los promotores fisiológicos ambientales o endógenos pueden desencadenar propiedades similares a las de las células madre/progenitoras en células preiniciadas que albergan mutaciones oncogénicas, actuando a través de diversas vías inflamatorias /vías de heridas a través de diferentes tejidos y tipos de células para alterar la selección clonal.
La lista de sospechosos que pueden actuar como promotores ambientales de tumores incluye, entre otros, microplásticos, glifosato(26), sustancias polifluoroalquiladas (PFAS), líquidos calientes(27) y agentes infecciosos como H. pylori (Figura 2).
Además, siguen sin respuesta serias preguntas con respecto a la seguridad a largo plazo de los cigarrillos electrónicos y la posibilidad realista de que la exposición humana a sustancias de vapeo pueda promover la iniciación de tumores, independientemente de la mutagénesis del ADN, de manera muy similar a como se cree que actúa la contaminación del aire.
Es probable que también desempeñen un papel formativo los factores endógenos o del estilo de vida, como la dieta, el estrés, la falta de sueño, el comportamiento sedentario y las alteraciones del ritmo circadiano, muchos de los cuales pueden afectar el microbioma.
Prevemos que definir enciclopedias de respuestas inflamatorias y otras respuestas reactivas inducidas por promotores ambientales (y fisiológicos) que orquestan la tumorigénesis facilitará el desarrollo de capacidades técnicas de importancia crucial para la detección temprana de neoplasias incipientes que puedan distinguir las lesiones que probablemente progresen a malignidad de aquellas que no lo harán.
Los ensayos rigurosos de promotores tumorales no mutagénicos requerirán capacidades tecnológicas mejoradas, incluida la secuenciación de ARN y ADN unicelular, así como transcriptómica/proteómica/metabolómica espacial, para revelar interacciones celulares heterotípicas y vías procesables adecuadas para los esfuerzos de prevención del cáncer molecular.
Si bien la base del cáncer puede residir en oncogenes mutantes y genes supresores de tumores, es cada vez más evidente que la progresión tumoral implica una programación epigenética no mutacional(4), como se refleja en la heterogeneidad dinámica aparente en muchos tumores y, como tal, en los epigenomas de ambos tipos de cáncer.
Las células y las diversas células del microambiente tumoral (TME) requerirán necesariamente iluminación en todas las etapas.
Existe evidencia de una “memoria epigenética” de exposición previa a agentes inductores de inflamación, que prevemos podría contribuir posteriormente a la tumorigénesis(28). Si bien la exposición pasada a mutágenos puede identificarse mediante la secuenciación del genoma completo de células y tejidos humanos que revela firmas mutacionales de carcinógenos distintivos(29), actualmente no existe ninguna tecnología para identificar la exposición pasada o actual a promotores tumorales.
El desarrollo técnico de tales herramientas permitirá los esfuerzos de prevención del cáncer molecular destinados a amortiguar las actividades promotoras de tumores inflamatorios, refinadas aún más por una profunda apreciación de las complejidades macroambientales más amplias del huésped, como se detalla en las secciones temáticas que siguen.
Tumorigénesis y cicatrización de heridas: dos caras de una misma moneda
Si bien la complejidad y singularidad de cada cáncer humano es innegable, también existen notables puntos en común entre los cánceres. Algunos de estos puntos en común son generales: todos los cánceres utilizan la misma maquinaria del ciclo celular y casi todos exhiben inactivación de la vía p53 y activación promiscua tanto de Myc como de la vía Ras/fosfatidilinositol 3-quinasa (PI3K).
Además, los cánceres de tipos o tejidos de origen específicos comparten fenotipos tumorales/estromales característicos, incluso cuando están impulsados por diferentes mutaciones oncogénicas, y estos difieren profundamente de los adenocarcinomas que surgen en otros órganos, incluso cuando están impulsados por las mismas mutaciones oncogénicas.
Dvorak nos ofreció la primera pista sobre el origen de estas limitaciones neoplásicas específicas de órganos con su propuesta de que los tumores son heridas no resueltas(30), y existen claros paralelismos entre las características distintivas del cáncer y las características de la cicatrización de heridas(31).
Más recientemente, esto ha sido confirmado directamente al mostrar que la activación de las mismas mutaciones oncogénicas centrales (KRasG12D y Myc) en tejidos adultos (pulmón y páncreas) impulsa directa e inmediatamente la formación de distintos adenocarcinomas cuyos fenotipos tumor-estromales coinciden perfectamente con los de sus homólogos espontáneos de adenocarcinoma de páncreas o pulmón humano.(32,33).
Por lo tanto, el principal determinante de los fenotipos del cáncer no es su panoplia única de mutaciones oncogénicas sino su órgano/tejido de origen. En el lenguaje contemporáneo, las mismas vías oncogénicas están pirateando los programas endógenos únicos de reparación de heridas de cada tipo de tejido/célula diana.
Este es un potente avance conceptual por dos razones. En primer lugar, habla de la presunción ampliamente extendida de que las características distintivas del cáncer (inflamación, supresión inmune, proliferación promiscua, supresión de la apoptosis, etc.) son rasgos neomórficos seleccionados a través de la evolución del tumor porque benefician la progresión del tumor(2).
Sin embargo, si los cánceres son simplemente ataques persistentes de los programas regenerativos normales, entonces la mayoría de las “características” del cáncer en realidad evolucionaron para optimizar la reparación del tejido, no un subterfugio neoplásico.
En segundo lugar, la reparación de heridas tiene componentes importantes: una fase regenerativa inicial, seguida de resolución, un programa morfogénico discreto que reorganiza el tejido involucrado y reafirma la arquitectura, la celularidad y la función homeostática.
La evidencia emergente indica que, en la cicatrización de heridas, el cambio de la regeneración a la resolución se desencadena por una disminución de la señalización mitogénica. Resulta provocativo que desde hace tiempo se sepa, gracias a estudios genéticos y farmacológicos, que el bloqueo de la señalización oncogénica desencadena una rápida regresión de los tumores (tanto de las células cancerosas como del estroma), al menos inicialmente.
Esto suele atribuirse a la “adicción a los oncogenes”, un fenómeno que carece de una explicación mecanicista coherente. Sin embargo, la similitud entre la resolución de la herida después de una lesión, debido al cese de la señalización mitogénica, y la regresión de los tumores tras el bloqueo oncogénico sugiere que un examen detenido de la resolución de la lesión podría generar estrategias completamente novedosas para el tratamiento del cáncer.
TME: desenmarañando y apuntando a nichos celulares
La inflamación inducida por los promotores del cáncer y la tumorigénesis de múltiples pasos resultante discutida anteriormente están entrelazadas con alteraciones específicas de la etapa en el complejo multicelular TME y su medio extracelular asociado, que coevoluciona progresivamente con las células cancerosas.
La evidencia emergente indica que las alteraciones en la integridad del tejido asociadas con la edad, la exposición al tabaco o los contaminantes ambientales, entre otros (ver la sección envejecimiento y cáncer: aptitud celular, dinámica del microambiente y evolución en el tiempo) pueden permitir expansiones clonales de células somáticas normales que albergan mutaciones oncogénicas.
Evidentemente, el TME participa en la regulación de la progresión de la enfermedad y en la modulación de la respuesta a una amplia gama de terapias contra el cáncer(31,34). Dentro del TME, los nichos celulares proporcionan hábitats únicos que influyen en el comportamiento del tumor, la respuesta al tratamiento y la vigilancia inmune.
Comprender las complejas interacciones dentro de estos nichos espaciales es esencial para desarrollar tratamientos contra el cáncer más eficaces. Además, existe la posibilidad de que al identificar y apuntar a los tipos de células, procesos o vías de señalización clave dentro de nichos particulares, se puedan amplificar los efectos de tales intervenciones terapéuticas. A continuación resumimos brevemente el conocimiento sobre los nichos celulares dentro del TME y destacamos preguntas que justifican la investigación durante las próximas décadas.
Los nichos celulares en el TME consisten en diversas poblaciones de células, incluidas células cancerosas, células inmunes, fibroblastos, vasculatura, grasa, células nerviosas y componentes de la matriz extracelular (MEC).
Cada nicho puede contribuir al crecimiento, la invasión y la metástasis del tumor de distintas maneras al respaldar las características específicas de las células cancerosas que los habitan (Figura 3).
Por ejemplo, las células cancerosas con un alto potencial de crecimiento residen dentro de nichos similares a las células madre(32), que apoyan su autorrenovación y resistencia a las terapias convencionales, lo que lleva a la recaída de la enfermedad(33,35).
Los nichos de células inmunes pueden promover o suprimir las células antitumorales. respuestas inmunitarias, lo que influye profundamente en los resultados del tratamiento(36). La heterogeneidad espacial de otros componentes de TME, como los fibroblastos en el estroma tumoral, puede contribuir a la desmoplasia, la angiogénesis y la modulación inmunitaria(37).
La importancia de los nichos de cáncer se puede apreciar particularmente en el contexto de la colonización metastásica, donde las células metastásicas deben recrear un entorno de apoyo al sembrar un nuevo órgano.
En particular, el hecho de que las metástasis puedan estallar años después de la resección del tumor primario indica que las células competentes para la metástasis pueden residir en órganos distantes en un estado latente y no proliferativo. Se informa sobre la existencia de componentes de nicho inactivos que mantienen el estado inactivo pero viable de las células metastásicas, con propensión a permitir el crecimiento(38,39). Sin embargo, las propiedades de estos nichos inactivos y cómo cambian con el tiempo para reactivar las células cancerosas son todavía en gran parte desconocidos.
Ciertamente, el envejecimiento de los tejidos tiene implicaciones para la reversión de la latencia(40). Como analizamos en las siguientes secciones, el envejecimiento, así como los cambios sistémicos progresivos que están directamente relacionados con las perturbaciones metabólicas e inflamatorias causadas por factores derivados del cáncer, pueden contribuir a la evolución de nichos (Figura 3).
Se han logrado importantes avances tecnológicos en la caracterización de nichos celulares dentro del TME, revelando su naturaleza dinámica y compleja.
Las técnicas de imagen avanzadas, la secuenciación unicelular, la transcriptómica espacial, las técnicas de imagen múltiple de alta resolución y las estrategias de etiquetado de nichos están permitiendo a los investigadores identificar distintas poblaciones celulares y mapear su organización espacial dentro de los tumores(41). Esto ha permitido, por ejemplo, la identificación de un programa regenerativo intrínseco del tejido activado en las primeras etapas del inicio del nicho metastásico(41), los estados fenotípicos inherentes a nichos tumorales particulares vinculados a ciertas anomalías genéticas o entre pacientes con cánceres primarios o metastásicos(42,43), los cambios en la composición del estroma tumoral asociados con la progresión del cáncer (44), así como las características en el TME que pueden correlacionarse con el resultado clínico o predecir las respuestas al tratamiento(45,46,47).
Con base en estos perfiles multiómicos, están surgiendo nuevas clasificaciones de tumores(48). Aunque esperamos que muchos tumores
En última instancia, será susceptible de modulación TME en el futuro, es posible que ciertos cánceres resulten especialmente desafiantes, como las metástasis hepáticas en pacientes con cánceres colorrectales estables a microsatélites o glioblastoma (GBM). En particular, estos también son órganos sujetos a supresión inmune en condiciones de estado estacionario, lo que probablemente contribuye a una barrera adicional para generar una respuesta inmune efectiva.
De cara al futuro, será importante determinar la biología subyacente a la diversidad espacial de TME; cómo la hipoxia, la disponibilidad de nutrientes, los metabolitos y la composición de la ECM influyen en el comportamiento de los nichos celulares.
Comprender la intrincada interacción entre los distintos nichos celulares dentro de los tumores es esencial, por ejemplo, para discriminar entre nichos que promueven el cáncer y los que lo restringen.
La investigación de la evolución temporal de estos nichos celulares proporcionará información sobre cómo evoluciona el TME tanto durante la progresión del cáncer como después de una intervención terapéutica.
Los estudios longitudinales que capturen la dinámica de las poblaciones celulares dentro de nichos a lo largo del tiempo, por ejemplo, a partir de biopsias secuenciales de pacientes combinadas con estrategias de imágenes multimodales en modelos preclínicos(49), serán clave para desentrañar la heterogeneidad espacio-temporal de los tumores.
Se puede prever que trazar la evolución dinámica del TME y sus nichos heterogéneos revelará nuevas vías para manipular los estados del TME para interferir con el crecimiento y la progresión del tumor, el crecimiento metastásico y la resistencia adaptativa a la terapia.
Comprender los determinantes clave de los nichos inactivos que pueden mantener las células metastásicas en un estado inactivo durante décadas, y las alteraciones posteriores que conducen a un cambio hacia el crecimiento metastásico, será un requisito esencial para desarrollar estrategias para detectar y eliminar las células inactivas. Generar sistemas modelo refinados para investigar estas cuestiones será fundamental para obtener conocimientos mecanicistas sobre la latencia metastásica y otros fenotipos específicos.
La explotación de las vulnerabilidades dentro de los nichos celulares tiene un gran potencial para el desarrollo de estrategias terapéuticas efectivas basadas en la precisión mediante la identificación de marcadores o vías específicas que son esenciales para el mantenimiento del nicho y la progresión del tumor. Más allá del TME está el “macroambiente” tumoral del huésped, también representado en la Figura 3, que abarca varias de las complejidades que se analizan en las siguientes secciones.
El macroambiente inmunológico: control homeostático y fisiología de los sistemas de piratería.
Los efectos de las perturbaciones del sistema inmunológico inducidas por tumores se extienden más allá del entorno inmunológico local del tumor, implicando alteraciones pronunciadas en el paisaje inmunológico sistémico durante la tumorigénesis.
Las moléculas paracrinas producidas por células cancerosas, células inmunes y células estromales no inmunes durante la progresión del tumor y liberadas en el sistema cardiovascular tienen acciones más allá del TME local (Figura 4).
Se han identificado varios mecanismos que conducen a la liberación de moléculas inmunomoduladoras durante la progresión del cáncer. Primero, el tumor en sí puede actuar de manera autocrina sobre células tumorales y no tumorales vecinas, lo que lleva a la activación de células tanto inmunes como no inmunes y a la liberación de moléculas adicionales que se suman al secretoma del TME.50.
Segundo, la senescencia celular Los programas que comúnmente son activos en células malignas y no malignas pueden exacerbarse con agentes quimioterapéuticos, lo que resulta en un fenotipo secretor marcado por una alta producción de citocinas proinflamatorias como la interleucina (IL)-6 y la IL-8(51,52,53, 54).
En tercer lugar, las células tumorales genéticamente inestables activan sensores de daño en el ADN que producen interferones tipo 1 o activan los inflamasomas y la producción de IL-1β, que pueden actuar periféricamente sobre las células estromales y los progenitores de la médula ósea(50,55,56,57,58).
En cuarto lugar, los microbianos. elementos influenciados por la dieta y el entorno de los pacientes pueden conducir a la activación de células que expresan receptores de reconocimiento de patrones y la liberación de moléculas inflamatorias como IL-6, IL-1β y factor de necrosis tumoral (TNF)(59,60,61,62,63,64,65).
Por último, los metabolitos inducidos por el estrés producidos por células tumorales genéticamente inestables o células inmunes y estromales circundantes también pueden liberarse activamente en el sistema circulatorio(66,67,68,69).
Estas señales sistémicas son detectadas por progenitores hematopoiéticos durante el curso del desarrollo del tumor, lo que conduce a una mielopoiesis desregulada y a la expansión de células mieloides aberrantes que contribuyen a la amortiguación de la inmunidad antitumoral local y sistémica, y a la formación de un TME protumoral (Figura 4)(70,71).
La cooptación de programas de desarrollo y regeneración por parte de las células tumorales contribuye aún más a los cambios en el entorno inmunológico sistémico, como lo ejemplifica la cooperación entre MYC y KRAS para impulsar un microambiente inmunosupresor, marcado por la exclusión de infiltrados sistémicos de células T y B en pacientes altamente inmunes.
La desactivación de MYC desencadena la regresión del tumor que se acompaña de la rápida reversión de estos cambios inmunológicos, lo que implica un programa que es paralelo a la fase de resolución de las respuestas de curación de heridas(72).
Además, las características de las respuestas inmunes sistémicas en huéspedes con tumores están fuertemente influenciadas por cambios en los controles homeostáticos fisiológicos, como el control circadiano, la respuesta endocrina al estrés y el metabolismo, y los cambios inducidos por la actividad física en el sistema circulatorio.
Comprender cómo cambia la respuesta inmune sistémica durante la progresión del cáncer, y cómo las funciones inmunes sistémicas se alteran a medida que aumenta la diversidad antigénica y fenotípica de un tumor, y si la inmunosupresión adaptativa provocada por la evolución del tumor contribuye a la muerte relacionada con el cáncer, son temas importantes para investigación futura.
Estas complejidades sugieren que el diseño de terapias inmunológicas efectivas requerirá una comprensión integral de los fundamentos moleculares de la interacción entre la inmunidad local y sistémica.
En términos generales, estos análisis comprenderán la identificación de los impulsores sistémicos de la mielopoiesis patógena a través de un perfil profundo y dinámico del secretoma de los pacientes con cáncer, así como el trazado de los programas de cromatina y transcriptómicos a lo largo del linaje mieloide, comenzando desde los progenitores hematopoiéticos tempranos en la médula ósea hasta células mieloides asociadas a tumores para identificar los ganglios iniciadores de la desregulación mieloide. Los resultados pueden guiar el desarrollo de nuevas estrategias para atacar los mediadores de la disfunción inmune sistémica.
Las evaluaciones simultáneas de los secretomas de los pacientes con cáncer sistémico y sus características clínicas, junto con una comprensión de la evolución somática del tumor, podrían formar la base de un marco terapéutico para guiar la inmunoestratificación del paciente y la toma de decisiones inmunoterapéuticas.
Es importante destacar que la tumorigénesis de varios pasos, la progresión maligna y los fenotipos en evolución de los microambientes y macroambientes sistémicos que promueven el cáncer también están modulados por una gran cantidad de otras complejidades, como se detalla a continuación.
Envejecimiento y cáncer: aptitud celular, dinámica del microambiente y evolución en el tiempo.
La mayoría de los cánceres ocurren en personas mayores de 60 años y, para 2050, se proyecta que más de 2 mil millones de la población mundial tendrá más de 60 años(73).
El aumento del cáncer relacionado con la edad puede atribuirse a varios factores, incluido el acumulación de daño genético crónico, deriva epigenética, alteraciones en los microambientes tisulares (incluido el aumento de células senescentes) y cambios en la inmunidad adaptativa e innata(74).
Estos factores pueden alterar la homeostasis del tejido y, por tanto, la aptitud, permitiendo la selección para la expansión proliferativa de células mutantes que responden a tales paisajes alterados.
Sin embargo, dada la frecuencia con la que se observan en tejidos normales, se puede suponer que las expansiones clonales impulsadas por mutaciones rara vez evolucionan hasta convertirse en neoplasias malignas potencialmente mortales en microambientes de tejidos envejecidos(75).
Es fundamental considerar la asociación de los cánceres con la vejez. A través de la lente de la biología evolutiva, los animales han desarrollado estrategias para mantener las funciones de los tejidos y evitar enfermedades, maximizando el éxito reproductivo.
Estos mecanismos disminuyen en los períodos post-reproductivos. Por lo tanto, debemos apreciar cómo los microambientes de tejido joven/sano maximizan la aptitud somática de las células madre y progenitoras, previniendo la persistencia o expansión de células con mutaciones potencialmente malignas: la juventud suprime los tumores.
Desafortunadamente, los tejidos envejecidos pierden progresivamente su capacidad de limitar la evolución del cáncer(75,76). En particular, los síndromes de progeria heredados a menudo (pero no siempre) exhiben una incidencia de cáncer acelerada y aumentada(77), que prevemos es el resultado de una disfunción tisular asociada a la progeria en el contexto de una mayor frecuencia de mutaciones.
Estudios recientes han revelado que el envejecimiento se caracteriza por un aumento de las expansiones clonales de células somáticas que albergan mutaciones oncogénicas en el tejido histológicamente normal(78), que pueden variar ampliamente en su potencial patogénico, como se ha demostrado más claramente en el caso de la hematopoiesis clonal, que se asocia con un aumento de riesgo de leucemias y cánceres sólidos(79).
Un desafío será comprender mejor cómo los tejidos sanos impiden la expansión clonal que daña los tejidos y la evolución fenotípica de las células somáticas que conduce a la patogénesis del cáncer, y cómo los carcinógenos ambientales y los factores del estilo de vida en conjunto con el envejecimiento natural afectan estos mecanismos protectores (consulte la sección tumorigénesis de múltiples pasos: comprensión de la promoción y progresión del tumor).
Estas expansiones están asociadas con condiciones que cambian el paisaje de los tejidos, incluida la enfermedad inflamatoria intestinal(80,81,82), la exposición al sol de la piel(83), el alcohol y el tabaquismo para el esófago(84), la contaminación del aire y la exposición al tabaquismo de los pulmones(21), y la obesidad y el tabaquismo para la hematopoiesis clonal(85).
Aunque cada vez se acepta más que los estilos de vida saludables promueven la reparación/mantenimiento de los tejidos y la protección contra el cáncer y que los estilos de vida poco saludables engendran inflamación crónica, mala reparación de los tejidos y un mayor riesgo de cáncer y otras enfermedades(86,87), no entendemos cómo los estilos de vida saludables afectan las primeras etapas de la carcinogénesis, incluidas las expansiones clonales impulsadas por mutaciones.
Los conocimientos futuros pueden respaldar el desarrollo de intervenciones que puedan bloquear las expansiones clonales anormales en los tejidos envejecidos, con el objetivo de reducir la incidencia del cáncer y el envejecimiento fisiológico.
Será fundamental comprender cómo los carcinógenos no mutagénicos (análogos al potencial de promoción de tumores de las partículas contaminantes) impactan este proceso e impulsan la expansión de células con potencial similar a un tallo que alberga mutaciones oncogénicas. También necesitamos caracterizar supuestos circuitos de retroalimentación mediante los cuales el envejecimiento promueve expansiones clonales determinadas impulsadas por mutaciones, que luego contribuyen aún más al envejecimiento del tejido (que a su vez promueve la oncogénesis).
El análisis de patrones mutacionales clonales en muestras de tejido (p. ej., sangre y tejidos epiteliales accesibles) de ensayos clínicos de candidatos a agentes antienvejecimiento y de individuos de diferentes edades, estilos de vida (p. ej., ejercicio) y dieta podría revelar estrategias para limitar la evolución clonal que puede provocar cáncer y deterioro del tejido asociado al envejecimiento(80,81).
El envejecimiento también influye en la MEC, un componente central del TME secretado por fibroblastos asociados a tumores y otras células no tumorales(76). La MEC modula el comportamiento de las células tumorales mediante mecanotransducción.
Los fibroblastos envejecidos liberan moléculas que inducen cambios significativos en las células tumorales, lo que afecta las vías de señalización, la respuesta de las especies reactivas de oxígeno (ROS) y el metabolismo,(74,76), así como la salida de la latencia proliferativa(40). De hecho, es posible que los procesos reguladores que aumentan
La selección de clones somáticos que portan mutaciones asociadas al cáncer durante el envejecimiento se superpone con aquellos que despiertan a las células del estado latente para instruir la colonización metastásica (ver la discusión anterior sobre nichos metastásicos latentes)(40,88). Si bien la senescencia de los fibroblastos puede estar involucrada en ambas expansiones proliferativas.
Es importante distinguir entre senescencia y envejecimiento, ya que la senescencia puede ocurrir a lo largo de la vida y tener efectos tanto protumorales como antitumorales, como se describe en la siguiente sección. Dirigirse a elementos procesables en el microambiente de los tejidos envejecidos, más allá de la senescencia, tiene el potencial de inhibir la metástasis y superar la resistencia a la terapia en pacientes ancianos con cáncer.
Esta búsqueda requerirá técnicas sofisticadas de bioingeniería, patología basada en inteligencia artificial y exploración de factores que se cruzan como el sexo biológico y el estrés con la edad. Específicamente, la transcriptómica espacial en capas con secuenciación y la reconstrucción y anotación 3D del TME utilizando técnicas de patología basadas en IA como CODA(89), análisis cuantitativo de inmunohistoquímica múltiplex utilizando técnicas como AstroPath(89,90), así como herramientas bioinformáticas para el análisis del comportamiento genético para predecir la comunicación célula-célula será fundamental para comprender cómo el envejecimiento de la TME afecta la expansión de las células con potencial similar a un tallo que conduce al inicio de tumores o al despertar de células metastásicas latentes(91).
El objetivo final es identificar agentes que induzcan a los paisajes tisulares a asumir estados fenotípicos que limitan todas las etapas de la evolución del cáncer, desde expansiones clonales tempranas hasta crecimientos metastásicos.
Las aplicaciones podrían variar desde la prevención temprana (por ejemplo, intervenciones que mantienen paisajes más jóvenes) hasta terapias (por ejemplo, terapias adyuvantes/neoadyuvantes que reducen el crecimiento metastásico) que pueden usarse para reducir la carga del cáncer y la mortalidad en una población que envejece rápidamente.
Entre ellos, aprovechar la creciente base de conocimientos y las ideas sobre las complejidades que conectan la senescencia celular (parcialmente relacionada con la edad) con el cáncer probablemente sentará las bases, como se analiza a continuación.
Senescencia y cáncer: transiciones de estado celular y enfoques terapéuticos.
La senescencia es una respuesta al estrés caracterizada por un cese estable de la proliferación, con cambios en la morfología celular, la expresión genética, los estados de la cromatina, así como un aumento de la secreción de citocinas. La identificación inequívoca de las células senescentes se complica por la falta de marcadores estándar del estado senescente.
Además, existen varios estados celulares estrechamente relacionados, como la inactividad, la latencia, la diapausa y las células persistentes y tolerantes a los fármacos (DTP), que comparten características con las células senescentes. Por ejemplo, el marcador de senescencia β-galactosidasa asociada a la senescencia también se expresa en algunas DTP.
Además, si bien el dogma en el campo de la senescencia es que la detención de la proliferación de las células senescentes es irreversible (lo que no es el caso de los estados relacionados mencionados anteriormente), cada vez hay más pruebas de que la senescencia también es reversible(92).
Múltiples firmas de expresión genética pueden identificar células senescentes, pero existe una variabilidad significativa en la expresión genética entre las células cancerosas senescentes derivadas de diferentes tejidos(93).
En las células cancerosas, la senescencia puede desencadenarse por estrés genotóxico (resultante de la quimioterapia o radioterapia; denominado «senescencia inducida por terapia»), estrés oxidativo o señalización mitogénica hiperactivada.
Una detención de la proliferación puede considerarse beneficiosa a corto plazo para la terapia contra el cáncer. Sin embargo, la persistencia de células cancerosas senescentes puede ser desfavorable a largo plazo debido a la creación de un microambiente inflamado, actuando así como promotor de tumores (ver sección promoción de tumores).
Es cada vez más factible, y potencialmente beneficioso, matar células cancerosas senescentes, fibroblastos y células normales afectadas colateralmente, mediante la llamada terapia senolítica, con el objetivo de evitar efectos fenotípicos no deseados de la senescencia en el cáncer.
De hecho, la evidencia de modelos de ratón sugiere que varios de los efectos secundarios de los agentes quimioterapéuticos son causados por la inducción de senescencia en células normales, de modo que la eliminación de células senescentes en ratones tratados con quimioterapia redujo la supresión de la médula ósea y mejoró la función renal(94).
Envejecimiento
Los estudios también han demostrado un aumento de células senescentes (principalmente fibroblastos) con el tiempo, y la eliminación senolítica de dichas células senescentes en modelos animales retrasa los trastornos asociados con el envejecimiento, incluido el cáncer (consulte la sección sobre envejecimiento)(95,96).
Con base en estas consideraciones, se ha propuesto un enfoque de “dos golpes” para la terapia contra el cáncer, que consiste en un tratamiento secuencial con una terapia inductora de senescencia, seguida de una terapia senolítica(92).
Conceptualmente, el tratamiento secuencial debe ser altamente sinérgico, al tiempo que reduce la toxicidades de la terapia combinada. Una complicación práctica en la aplicación de este enfoque es la heterogeneidad de las vías de senescencia de las células cancerosas, lo que dificulta encontrar fármacos senolíticos que actúen de manera amplia.
Por ejemplo, el fármaco mimético BH3 navitoclax (ABT-263), que se utiliza ampliamente como fármaco senolítico en la investigación del envejecimiento, sólo es senolítico en una fracción de las células cancerosas senescentes.
Datos recientes indican que la activación de la señalización del receptor de muerte con anticuerpos agonistas tiene una actividad senolítica más amplia en las células cancerosas(97). Un enfoque alternativo a la senólisis es la explotación de las células inmunitarias atraídas por la masa tumoral senescente mediante citocinas secretadas que son componentes de la senolisis asociada programa secretor (SASP).
En modelos preclínicos de cáncer de páncreas, la inducción de la senescencia resultó en el reclutamiento de células T CD8+ en los tumores, lo que resultó en sensibilidad a la inmunoterapia de punto de control(98).
A pesar de estos avances iniciales, aún quedan muchas preguntas por responder. La explotación exitosa del enfoque terapéutico prosenescencia de “un doble golpe” requerirá una comprensión de qué combinaciones de fármacos prosenescencia más senolíticos son más activas y del contexto de dependencia de dichos pares de fármacos. Además, necesitamos comprender mejor la complejidad de las células inmunitarias que se infiltran en los tumores senescentes y cómo explotar su presencia terapéuticamente.
Sin embargo, existe otra capa de complejidad, a saber, que mantener las células senescentes puede ser beneficiosa en ciertos tumores, tal vez reflejando variabilidades en su SASP4; Esta dicotomía de función requiere más investigación con respecto al ajuste terapéutico de la senescencia.
Finalmente, en el cerebro humano que envejece, las células neuronales expresan abundantemente marcadores de senescencia(99).
Por lo tanto, será crucial comprender si se puede definir una ventana terapéutica para pacientes ancianos con cáncer en el contexto de la terapia senolítica.
Un estudio clínico de prueba de concepto exitoso que utilice terapias senolíticas y prosenescencia actuará como catalizador para este enfoque de la terapia contra el cáncer. Entrelazados con el envejecimiento y la senescencia están los efectos complejos de la variación metabólica, en las células cancerosas, en los tumores y en el huésped, como se detalla a continuación.
Metabolismo, dieta y cáncer: efectos sistémicos y oportunidades terapéuticas.
La desregulación generalizada del metabolismo y la energía celular en el cáncer sugiere que el metabolismo alterado no es simplemente una consecuencia auxiliar de la tumorigénesis, sino más bien un requisito seleccionado para el inicio y la progresión del tumor.
Si bien los cambios metabólicos intrínsecos a las células cancerosas apoyan la tumorigénesis, la compleja interacción metabólica entre el tumor y el huésped, tanto dentro del TME inmediato como a través de órganos distantes, resalta nuestra creciente apreciación del cáncer como una enfermedad sistémica (Figura 5).
Temprano durante la carcinogénesis, las células cancerosas se seleccionan para una reprogramación metabólica que maximiza la producción y el suministro de nutrientes del microambiente para satisfacer las demandas metabólicas requeridas para un crecimiento aberrante(100).
Sin embargo, la relación recíproca entre el metabolismo tumoral y el metabolismo sistémico permanece en gran medida inexplorada. En esta sección nos centramos en nuestra creciente apreciación de estas complejidades entrelazadas y su potencial para proporcionar nuevos enfoques para prevenir, detectar y tratar el cáncer.
Efectos del cáncer en el cuerpo.
Las células cancerosas subvierten las células no cancerosas (p. ej., fibroblastos, adipocitos, células inmunitarias y neuronas) en el TME para proporcionar apoyo metabólico y complementar los entornos tumorales pobres en nutrientes(101,102), en los que la ávida absorción de nutrientes y la producción de desechos metabólicos pueden suprimir el sistema inmunológico anticancerígeno. respuestas(103,104,105).
En el macroambiente, los tumores alteran el metabolismo sistémico del huésped tanto directamente(102) (mediante la secreción del tumor de moléculas de señalización particulares o la alteración de la disponibilidad de nutrientes) como indirectamente(106) (por ejemplo, a través de la respuesta del sistema inmunológico al tumor).
La reprogramación del metabolismo en órganos distantes, como el hígado(107) y el cerebro(108), altera el metabolismo de todo el cuerpo para promover manifestaciones sistémicas relacionadas con el cáncer, incluidas metástasis, resistencia a la terapia contra el cáncer, caquexia asociada al cáncer (CAC) y muerte ( Figura 5)(109,110).
Dado que estas alteraciones en el metabolismo sistémico comienzan temprano, comprenderlas puede guiar y permitir el diseño de estrategias destinadas a preservar el metabolismo del huésped y restringir las manifestaciones sistémicas de los cánceres en etapa tardía.
Un ejemplo claro del efecto metabólico del cáncer en el cuerpo es la inducción del estado catabólico activo de CAC, un síndrome de emaciación complejo y debilitante que limita el estado físico y la efectividad de las terapias, lo que resulta en malos resultados para los pacientes(111,112).
El CAC probablemente representa una corrupción de un proceso normal y transitorio de cicatrización de heridas, que no puede resolverse en el contexto del cáncer(113). Los mecanismos por los cuales la progresión tumoral y la metástasis causan cambios sistémicos que facilitan la pérdida de músculo y/o grasa siguen siendo en gran medida desconocidos, pero pueden reflejar una relación cáncer/huésped, interacción que provoca un desequilibrio metabólico sistémico que favorece al tumor a expensas del huésped.
En consecuencia, las intervenciones que previenen o revierten la CAC también pueden limitar el crecimiento tumoral(114). Una comprensión más profunda de los distintos impulsores metabólicos, inflamatorios, moleculares y neuroendocrinos de la CAC y sus orígenes en el huésped o en el tumor en evolución puede abrir nuevas vías terapéuticas que se extiendan más allá de tratar las causas y consecuencias de la CAC, hasta obstaculizar el crecimiento del tumor primario y metastásico(114).
Obesidad, actividad física y cáncer.
Los trastornos metabólicos, así como las variaciones en los estados fisiológicos saludables afectados por factores como el ejercicio, pueden influir en el desarrollo y la progresión de los cánceres y es probable que impliquen una interacción entre el genoma, la dieta, el estado físico, el estado fisiológico y el microbioma del huésped(115).
Aproximadamente el 4 % a 8% de todos los cánceres se atribuyen directamente a la obesidad(116), pero esta estadística aumentará rápidamente a medida que crezcan las tasas de obesidad, particularmente en los países de ingresos bajos y medianos.
Aunque las estrategias para reducir la obesidad y aumentar la actividad física regular son intervenciones obvias con potencial para prevenir o tratar el cáncer, el vínculo mecánico entre la obesidad y el cáncer sigue sin estar claro.
Las preguntas pendientes incluyen las siguientes: ¿es la obesidad per se o la perturbación metabólica que la acompaña responsable del aumento de la incidencia del cáncer? ¿La expansión de los adipocitos libera indiscriminadamente factores prooncogénicos? ¿Tienen algunos individuos una respuesta fisiopatológica específica a la obesidad que los haga más susceptibles al cáncer? ¿Y cuál es el papel del microbioma intestinal (que se analiza con más detalle a continuación)?
La actividad física regular ha sido implicada en la prevención y/o mejora de la supervivencia específica del cáncer para varios tipos de cáncer(117), aparentemente funcionando no sólo reduciendo el índice de masa corporal sino también disminuyendo los niveles de hormonas, suprimiendo la inflamación, mejorando la función inmune y alterando los niveles de hormonas intermedias y metabolismo(118). La aparición de nuevas tecnologías para identificar efectores moleculares de la actividad física a nivel del organismo(119,120) brindará oportunidades sin precedentes para definir respuestas fisiológicas a la actividad física que pueden aprovecharse para la prevención y el control del cáncer.
Cáncer y metabolismo del huésped en detección y terapia.
Las intervenciones metabólicas que afectan las células cancerosas, el TME o el metabolismo sistémico del huésped (mediante la limitación del apoyo nutricional de los cánceres o el aumento de las respuestas antitumorales del huésped) pueden brindar nuevas oportunidades terapéuticas.
Sin embargo, atacar las alteraciones metabólicas en el cáncer ha demostrado ser un desafío, debido a muchos factores, incluidas las posibles toxicidades de las vías de ataque que son esenciales en las células normales y la dificultad de fabricar fármacos selectivos para enzimas muy abundantes, así como la flexibilidad y plasticidad metabólicas de células cancerosas.
Se necesitarán nuevos enfoques para superar estas limitaciones, incluidas estrategias para influir en las actividades patológicas de las enzimas metabólicas manteniendo al mismo tiempo su función en los órganos normales(121).
La capacidad de los cánceres para alterar el metabolismo sistémico y específico de los tejidos también sugiere nuevas oportunidades para la detección temprana de la enfermedad a través de cambios en los niveles o tipos de metabolitos circulantes o excretados(122).
Metabolismo redox, metabolismo del ciclo de la urea, metabolismo del azufre (incluida la producción de ácidos biliares), aspectos del metabolismo de un carbono y de nucleótidos, y posiblemente incluso de la función del ciclo del ácido tricarboxílico (TCA) o de la glucólisis, podrían monitorearse sistemáticamente utilizando tecnologías cada vez más refinadas para informar sobre el metabolismo del huésped, el tumor y su interacción.
Las intervenciones dietéticas son otra vía prometedora, con muchas probabilidades de ser efectivas para mejorar la terapia contra el cáncer(123,124).
El ayuno o las dietas que imitan el ayuno (FMD, por sus siglas en inglés) pueden mejorar los resultados del cáncer para muchos tumores específicos de órganos tanto en roedores como en humanos(125,126), evidentemente funcionan a través de la regulación de varias vías reguladoras, incluida la reducción de la señalización de insulina y leptina, y la inflamación atenuada (que también se regulan negativamente con el ejercicio).
La nutrición de precisión (alteraciones selectivas de carbohidratos, aminoácidos y lípidos específicos de la dieta) ofrece otra forma de abordar las vulnerabilidades al cáncer. Reducir la ingesta de aminoácidos no esenciales como la serina, la glicina y la metionina puede retardar el crecimiento tumoral y sensibilizar las células cancerosas resistentes al tratamiento(127,128).
Además, manipular el equilibrio de los lípidos saturados e insaturados puede afectar directamente la composición de la membrana y, por tanto, perjudicar la supervivencia. de las células cancerosas(129).
Además, las dietas bajas en carbohidratos que limitan la producción de insulina pueden aumentar la eficacia de las terapias dirigidas a PIK3CA en ciertos cánceres(130).
Si bien varios de estos enfoques dietéticos se están explorando ahora en entornos clínicos, es necesario comprender mejor cómo la dieta afecta la normalidad. Las respuestas fisiológicas y anticancerígenas aún están surgiendo(123,124,30) y se puede anticipar que guiarán futuras aplicaciones de esta estrategia.
El éxito de tales intervenciones metabólicas depende de una comprensión mecanicista más profunda de los requisitos impuestos a las células cancerosas por su origen, genética y entorno, junto con el impacto de la terapia(123,131).
También necesitamos comprender la interacción y los efectos del metabolismo y sus efectos fisiológicos. y modulaciones farmacológicas en muchos de los factores sistémicos del huésped que gobiernan la progresión del cáncer, como se analiza en otra parte de esta revisión, incluidos los ritmos circadianos, como se analiza a continuación.
Ritmos circadianos y cáncer: los relojes como reguladores críticos de la fisiología y la enfermedad
Un aspecto poco comprendido de la dinámica tumor-huésped implica los posibles efectos temporales sobre el comportamiento de las células cancerosas y la respuesta a la terapia.
Los ritmos circadianos describen oscilaciones de luz, temperatura y otros aspectos del entorno terrestre durante 24 horas, así como fluctuaciones diarias en la expresión genética y la fisiología impulsadas por osciladores moleculares codificados genéticamente («relojes circadianos») presentes en prácticamente todas las células de los mamíferos.
La alteración de los ritmos circadianos causada por el trabajo nocturno, los viajes a través de múltiples zonas horarias y la residencia en el borde occidental de las zonas horarias se asocian con una mayor incidencia de cáncer(132). Trabajos recientes han descubierto mecanismos que pueden contribuir a estas asociaciones.
Los relojes circadianos de los mamíferos se basan en un circuito de retroalimentación de transcripción-traducción en el que un heterodímero de los factores de transcripción CLOCK y BMAL1 impulsa la expresión de sus represores, PERÍODOS (PER1–3) y CRIPTOCROMOS (CRY1 y 2).
Los PER y CRY se acumulan e inactivan CLOCK-BMAL1, después de lo cual los PER y CRY se marcan para la degradación proteasomal, renovando el ciclo. Miles de genes exhiben oscilaciones diarias de expresión en cada órgano examinado, de modo que más de la mitad del genoma se expresa rítmicamente en algún lugar del cuerpo(133).
Varios mecanismos contribuyen a las oscilaciones transcripcionales, incluida la transactivación por CLOCK-BMAL1 y la represión de diversas transcripciones. factores por PER y CRY. Curiosamente, se ha demostrado que tanto PER2 como CRY2 influyen en P53, el supresor de tumores inactivado con mayor frecuencia en el cáncer(134,135). CRY2 también estimula el recambio de la oncoproteína c-MYC en células que proliferan rápidamente(136), aunque su eliminación no parece afectar a MYC en personas sanas(137).
Las conexiones moleculares entre los relojes y las proteínas que se sabe que influyen en el cáncer, como P53 y c-MYC, alientan la especulación de que la interrupción de estas vías podría explicar una mayor incidencia de cáncer asociada con anomalías circadianas.
Sin embargo, trabajos recientes sugieren mecanismos alternativos, incluida la alteración de la inmunidad antitumoral y de las respuestas al estrés celular(138,139).
La eliminación genética de componentes individuales del reloj tiene impactos variados sobre el crecimiento tumoral en modelos de ratón(132,140,141), por lo que parece poco probable que se pueda aplicar un mecanismo particular basado en la alteración de la inmunidad antitumoral y de las respuestas celulares al estrés(138,139). Un componente central del reloj explicará universalmente el aumento de la incidencia de cáncer causado por la alteración circadiana.
El uso de células murinas transformadas para iniciar tumores produce diferencias sorprendentes en el crecimiento tumoral dependiendo de la hora del día en la que se implantaron las células(139), lo que implica una regulación circadiana de las respuestas inmunes antitumorales.
La exposición de los ratones huéspedes a la alteración circadiana crónica exacerbó el crecimiento del tumor y eliminó el impacto del tiempo de injerto, lo que sugiere que las respuestas inmunes alteradas pueden contribuir a un mayor crecimiento del tumor tras la alteración circadiana.
La alteración circadiana crónica también aumentó la carga tumoral en modelos de ratón genéticamente modificados de adenocarcinoma de pulmón impulsado por KRAS(138,142). Un análisis imparcial de la expresión génica en tumores de pulmón reveló que la alteración circadiana aumenta la expresión del factor de choque térmico 1 (HSF1), que se ha relacionado con una mayor tumorigénesis en una variedad de contextos(143).
Se necesita investigación adicional para determinar si se requiere una vigilancia inmune reducida y/o HSF1 elevado para mejorar la tumorigénesis causada por la alteración circadiana crónica.
Es probable que múltiples mecanismos contribuyan a la mejora de la tumorigénesis observada en personas expuestas a alteraciones circadianas.
La alteración circadiana también influye en los fenotipos asociados al cáncer, incluidas la metástasis y la caquexia(144,145). La alteración circadiana actúa como un factor de estilo de vida que promueve tumores a través de mecanismos desconocidos, que probablemente incluyen la privación del sueño y las alteraciones metabólicas.
Es necesario continuar la investigación para delinear las contribuciones de los mecanismos discutidos aquí y los efectos adicionales de la alteración circadiana en la etiología de diversos tipos de tumores. Pocos ensayos clínicos incluyen múltiples tiempos de dosificación y una “lógica circadiana” en su diseño, aunque hay indicios de que dichos regímenes pueden afectar los resultados de la quimioterapia(146), y la respuesta al bloqueo de los puntos de control en el melanoma, donde la evidencia emergente del estudio MEMOIR sugiere que las respuestas inmunes adaptativas son menos sólidos por las noches(147).
Registrar la hora del día en que se recolectan las biopsias y cuándo se administran los tratamientos durante los ensayos clínicos también mejoraría notablemente el potencial para avanzar en la comprensión en esta área. Otro regulador sistémico de la interacción entre los fenotipos del huésped y del tumor, probablemente entrelazado con los ritmos circadianos, involucra al sistema nervioso, como se analiza más adelante.
Influencias del sistema nervioso: roles emergentes de la diafonía entre neuro y cáncer
Neuronas, células gliales y nervios: un componente crítico del ecosistema del cáncer
En los últimos años, tanto el sistema nervioso central como el periférico se han convertido en componentes críticos de las interacciones tumor-huésped.
Descubrimientos apasionantes han establecido la inervación neuronal y la subversión de la señalización como nuevos factores potenciales del macroambiente y microambiente tumoral, tanto en los tumores cerebrales como en la mayoría de los otros tipos de cáncer, lo que hace posible que las interacciones entre las neuronas y el cáncer eventualmente se conviertan en otro sello distintivo del cáncer.
Por mucho que gobierne la cicatrización fisiológica de heridas, el desarrollo de tejidos y la organogénesis, así como la homeostasis y la plasticidad a lo largo de la vida, el sistema nervioso parece desempeñar papeles instrumentales en la regulación de los cánceres(5,148).
Específicamente, la actividad neuronal sináptica y paracrina, pero también la actividad oncológica, las características neuronales intrínsecas pueden regular el inicio, el crecimiento, la diseminación y la resistencia al tratamiento del cáncer; por el contrario, los tumores pueden afectar negativamente al sistema nervioso e incluso reprogramarlo, lo que genera circuitos de retroalimentación perjudiciales(5,148). Además, el sistema nervioso influye en la biología del cáncer y la respuesta a la terapia contra el cáncer a través de la desregulación del sistema inmunológico, alteraciones de la angiogénesis y efectos sistémicos más amplios.
Un ejemplo destacado es el GBM (gliomas), en el que las vías del desarrollo neurológico normal son secuestradas para interconectar las células cancerosas en una red, un sincitio que involucra comunicaciones entre células que fomentan los fenotipos tumorales(149).
Esta red de cáncer recibe información directa de células glutamatérgicas (excitatorias) genuinas. sinapsis neurona-glioma que activan la red, impulsando el crecimiento del tumor(150). La población celular es estimulada constantemente por una actividad rítmica autónoma generada por células cancerosas similares a marcapasos dentro de la red, recapitulando los procesos de desarrollo neurológico(151).
Diseminación del GBM (invasión perineural) en el cerebro está gobernado por esta red de cáncer a través de sus asociaciones neurona-glioma, así como por otros mecanismos neuronales cooptados(152).
Sorprendentemente, en el cerebro humano, GBM también remodela circuitos neuronales, como en regiones de representación del lenguaje, desarrollando interconectividad funcional entre el cáncer y el cerebro normal, lo que se asocia con una menor supervivencia del paciente(153).
Fuera del cerebro, los tumores secretan factores de crecimiento (p. ej., factor de crecimiento nervioso, NGF) que atraen la inervación de los nervios periféricos e incluso pueden reprogramarlos.
Se ha demostrado que esta inervación por los nervios simpáticos, parasimpáticos y sensoriales impulsa el crecimiento tumoral, la invasión, la metástasis y la resistencia terapéutica en la mayoría de los sitios de cáncer en todo el cuerpo, en gran medida a través de la secreción paracrina de factores neurales. Actualmente se están explorando nuevas vías terapéuticas basadas en estas interacciones neuro-cáncer(148).
Dolor en el cáncer
Un ejemplo importante de cómo los tumores y las terapias contra el cáncer pueden afectar negativamente al sistema nervioso de un paciente es la sensación de dolor.
El dolor óseo debilitante asociado con metástasis en los cánceres de próstata, mama y pulmón puede afectar gravemente la calidad de vida. Los analgésicos opioides siguen siendo el pilar del tratamiento del dolor del cáncer, pero las consecuencias a largo plazo de la tolerancia, la dependencia y la hiperalgesia son problemáticas(154).
La potente lidocaína analgésica tiene actividad analgésica y antitumoral, así como efectos sobre las respuestas inmunitarias, consistentemente con un papel vital para el eje neuroinmune del cáncer en los enfoques de tratamiento(155). Los ensayos clínicos de nuevos enfoques para el tratamiento del dolor centrados en bloquear la entrada al sistema nervioso central son prometedores, por ejemplo, el uso de anticuerpos que neutralizan mediadores derivados del sistema inmunológico como NGF o TNF para aliviar el dolor del cáncer . De manera similar, también se están evaluando pequeñas moléculas bloqueadoras de canales en ensayos de fase 3 destinados a reducir el dolor asociado al cáncer(156).
El control del dolor es una dimensión cada vez más importante para el manejo y tratamiento del cáncer, lo que resulta prometedor.
El camino hacia la traducción
Las investigaciones futuras en neurociencia traslacional del cáncer deberán abordar tres puntos principales: (1) mapear el mundo de las interacciones neurológicas-cáncer (por ejemplo, sinápticas, paracrinas y sistémicas), incluidas las clases de neurotransmisores, neurotrofinas, neuropéptidos y hormonas que son relevante para distintas entidades tumorales y etapas de la enfermedad, combinando imágenes moleculares, biomarcadores circulantes y perfiles morfológicos, neurofisiológicos y moleculares de tejidos tumorales recién resecados, para los cuales es necesario desarrollar metodologías personalizadas; (2) desarrollar biomarcadores informativos para monitorear las interacciones neuro-cáncer en un paciente determinado; (3) establecer socios óptimos de terapia combinada, por ejemplo, inmunoterapias, como parte del campo emergente de neuroinmunooncología.
Dado que hay más de 100 fármacos aprobados en neurología, psiquiatría y medicina interna que se dirigen a las vías de señalización neuronal, la reutilización de fármacos puede resultar productiva, como lo demuestran los ensayos clínicos dirigidos a las sinapsis estimuladoras de neuronas cancerosas del subtipo de receptor AMPA con el fármaco antiepiléptico perampanel(150,157), o apuntar a redes de células cancerosas de tipo neuronal con meclofenamato(149).
Además, el desarrollo de fármacos dirigido a reguladores tempranos del desarrollo neurológico usurpados por las células cancerosas y a interacciones neuronales específicas del cáncer presenta vías prometedoras.
Si bien interferir con la neurotransmisión normal puede provocar efectos secundarios que deben controlarse, comprender los mediadores y receptores específicamente implicados en impulsar la tumorigénesis debería producir objetivos terapéuticos basados en conocimientos mecanicistas más precisos.
En resumen, el reciente descubrimiento de interfaces íntimas entre el sistema neurológico y el cáncer abre nuevos horizontes en nuestra comprensión más amplia del cáncer, incluida la posibilidad de que la modulación de estados cognitivos u otros estados neuronales pueda alterar fenotipos cruciales del cáncer. Las investigaciones futuras determinarán si este conocimiento emergente puede aprovecharse para el desarrollo de nuevas estrategias terapéuticas.
Paridad: lecciones para la protección tumoral
Los ejemplos antes mencionados de cómo el huésped puede modular el desarrollo tumoral y la progresión maligna se han centrado en gran medida en las intersecciones que promueven los tumores.
La consideración contraria es el potencial del comportamiento humano y las exposiciones ambientales para proteger contra la iniciación de tumores.
Trabajos recientes han arrojado luz sobre el papel de las hormonas sexuales como los andrógenos y el sistema endocrino en tales interacciones y han revelado los mecanismos a través de los cuales gobiernan la inmunidad antitumoral y las respuestas de los puntos de control de las células T.
Esta interferencia es consistente con el conocimiento sobre la autoinmunidad y las enfermedades infecciosas, donde, por ejemplo, el sexo biológico se asocia con diferencias aparentes en la incidencia y magnitud de las respuestas inmunes mediadas por vacunas.
Un claro ejemplo de esto es la paridad y la duración de la lactancia materna, que están bien establecidos por tener beneficios positivos para la salud tanto de la madre como del niño.
El embarazo y la lactancia materna se han asociado con la protección contra el cáncer de mama y de ovario, aunque el riesgo aumenta en el corto plazo y un menor riesgo de mortalidad por todas las causas en el futuro(158).
El riesgo de cáncer de mama se reduce en un 4% por cada 12 meses de lactancia materna, además de la disminución del 7% en el riesgo observado en cada nacimiento(159). La lactancia materna reduce particularmente el riesgo de cánceres de mama triple negativos (TNBC)(160).
Se cree que los mecanismos detrás de la protección contra el cáncer de mama asociados con el embarazo están relacionados con la maduración de las células epiteliales mamarias, haciéndolas menos susceptibles a la transformación, y a una reducción de los estrógenos circulantes a través de la amenorrea.
Estudios anteriores han establecido un efecto pronóstico positivo de los altos niveles de infiltración de células T en cánceres de mama en etapa temprana, particularmente en TNBC(161).
Por ejemplo, se demostró que un pequeño subconjunto de células T con un fenotipo residente en tejidos era altamente proliferativo y citotóxico , cualidades esenciales para una respuesta antitumoral duradera(162).
Recientemente, utilizando modelos de ratón, se ha informado del papel crítico de las células T con un fenotipo de memoria residente (TRM) en la protección inmune contra la nueva exposición al tumor mamario, lo que sugiere que las TRM pueden conferir protección contra cáncer de mama(163).
El tejido mamario sano y no afectado por cáncer contiene una multiplicidad de células inmunitarias, incluidas células mieloides, asesinas naturales (NK), B y T que no expresan altos niveles de moléculas de puntos de control inhibidoras de células T(162,164).
Dado estos datos epidemiológicos, el trabajo futuro debería buscar evidencia de un mecanismo inmunológico que pueda explicar la relación entre la lactancia materna, el embarazo y la protección resultante contra el cáncer de mama a largo plazo.
Los patrones de expresión genética en el seno humano cambian significativamente años después del embarazo, en particular con una regulación positiva de genes relacionados con el sistema inmunológico(165).
El embarazo en sí causa cambios dramáticos en el microbioma intestinal y vaginal de la madre durante los tres trimestres(166).
También está bien aceptado que la leche materna contiene un rico microbioma que cambia a lo largo de las etapas de la lactancia(167). En las muestras de calostro, Weisella, Leuconostoc, Staphylococcus, Streptococcus y Lactococcus son las especies predominantes.
Por el contrario, al mes y a los 6 meses, los microbios que normalmente se encuentran en la cavidad bucal (p. ej., Veillonella, Leptotrichia y Prevotella) aumentan significativamente en la leche materna(168). En general, la leche materna puede contener hasta 600 especies bacterianas diferentes, que en última instancia, podría influir en el repertorio inmunológico tanto de la madre como del niño(169).
La investigación sobre los cambios inflamatorios y microbianos durante el embarazo y el postparto, así como la composición de la leche materna, se ha centrado principalmente en los beneficios para el lactante, pero aún no se ha investigado en profundidad cómo estos efectos pueden beneficiar a la madre a largo plazo, como así como contribuir a la vigilancia inmune del microambiente local de la mama.
Investigaciones adicionales sobre las hormonas sexuales, sus cambios en diversas etapas reproductivas, tanto en mujeres como en hombres, y cómo alteran la inmunidad antitumoral pueden ayudar a dilucidar los mecanismos moleculares subyacentes que gobiernan la protección y la progresión del cáncer, no solo en los órganos que responden a las hormonas.
Tromboinflamación y cáncer.
El tromboembolismo es la obstrucción de un vaso sanguíneo por la formación patológica de un coágulo. Es una de las principales causas de muerte en personas con cáncer y se asocia con un mal pronóstico que no puede explicarse por el estadio del cáncer(170,171).
Las personas con cáncer tienen un riesgo 9 veces mayor de sufrir trombosis que la población general y ;a incidencia del tromboembolismo asociado al cáncer está aumentanda (172).
Una predisposición subyacente a la trombosis se relaciona con una respuesta tromboinflamatoria desregulada que involucra la interacción entre la coagulación y los mediadores inflamatorios (Figura 6)(173).
Los neutrófilos, los leucocitos mieloides más abundantes, promueven la tromboinflamación mediante la liberación de trampas extracelulares de neutrófilos (NET), un efecto negativo combinación cargada de histonas, enzimas proteolíticas y ADN que activa la trombosis(174).
También se sabe que varios tipos de cáncer secretan factor tisular, el principal activador de la coagulación in vivo. Los niveles plasmáticos más altos de factor tisular se asocian con un peor estadio del cáncer, peores resultados y riesgo de trombosis(175).
La tromboinflamación promueve potencialmente el crecimiento y la diseminación del tumor. Se supone que los agregados de células tumorales y plaquetas promueven la migración del tumor(176).
Un recuento elevado de plaquetas es un marcador de mal pronóstico, así como de riesgo de tromboembolismo.
Los modelos murinos de GBM demuestran que las plaquetas se unen a las células tumorales a través de la podoplanina y, lo que es más importante, los niveles plasmáticos elevados de podoplanina se asocian con un mayor riesgo de tromboembolismo y menores tasas de supervivencia en los pacientes(176,177).
Los estudios en ratones y humanos han establecido asociaciones entre la cantidad y el tipo de neutrófilos intratumorales y la progresión de la enfermedad(178). El fenotipo de los neutrófilos está influenciado por el medio ambiente y muchos tipos de cáncer provocan potentes funciones inmunosupresoras y proangiogénicas de los neutrófilos que promueven el crecimiento tumoral y/o diseminación(179,180). Para equilibrar esta visión, la evidencia emergente también revela actividades antitumorales de los neutrófilos y factores derivados de tumores que actúan promoviendo la formación y activación de células T citotóxicas(181,182).
Más allá de estas funciones, se ha demostrado que la liberación de NET promueve el atrapamiento intravascular de células tumorales, lo que facilita la metástasis, al proteger físicamente a los tumores de las acciones de las células T citotóxicas o al despertar células cancerosas latentes al remodelar su microambiente local (Figura 6)(183).
Por el contrario, los NET también pueden ser citotóxicos y se ha demostrado que destruyen las células tumorales mediante la acción de enzimas proteolíticas e histonas asociadas.
Manejo de la trombosis
Muchos factores pueden alterar el riesgo trombótico durante el tratamiento de pacientes con cáncer, incluida la cirugía, la quimioterapia, la infección y la inoculación intravenosa de fármacos terapéuticos. Se necesita un estudio de cohorte a gran escala que mida en serie múltiples biomarcadores en personas con cáncer que reciben terapia para ayudar a identificar biomarcadores de riesgo que podrían usarse para definir un cambio en el riesgo de un individuo a lo largo del tiempo.
El desarrollo futuro de ensayos debería buscar marcadores globales fiables de riesgo trombótico que permitan un enfoque más flexible y personalizado. Esto nos lleva a la pregunta: una vez que podamos identificar el riesgo, ¿podemos hacer algo para mitigarlo?
El uso de anticoagulación en dosis bajas (tromboprofilaxis) para prevenir la tromboembolia venosa es eficaz en poblaciones de mayor riesgo de cáncer. Sin embargo, no hay evidencia suficiente para respaldar el uso de tromboprofilaxis en todas las personas con cáncer debido al riesgo de sangrado que provoca este tratamiento.
Las puntuaciones clínicas, la más validada es la puntuación de Khorana, pueden identificar a algunos, pero no a todos, en mayor riesgo(184).
Actualmente se están probando anticoagulantes más nuevos, como los inhibidores del factor XI, cuyo objetivo es “desacoplar” la hemostasia de la trombosis para el tratamiento de la trombosis por cáncer . Los futuros ensayos controlados aleatorios deberían investigar su eficacia para prevenir la trombosis.
Para mejorar los resultados para las personas con cáncer, necesitamos comprender el papel de la tromboinflamación y desarrollar métodos para monitorear el riesgo trombótico durante el manejo del cáncer a fin de mejorar las estrategias de prevención y tratamiento.
La intersección de la hematopoiesis clonal en la inflamación, la trombosis y la progresión tumoral.
Las mutaciones somáticas en las células madre hematopoiéticas están relacionadas con una mayor formación de NET y la promoción de la trombosis, tanto en el contexto de neoplasias malignas hematológicas como de la hematopoiesis clonal (p. ej., JAK2V617F en neoplasias mieloproliferativas o en la hematopoiesis clonal)(185).
La hematopoiesis clonal de potencial indeterminado (CHIP) es un fenómeno del envejecimiento definido por la presencia de mutaciones somáticas asociadas a la leucemia en las células madre hematopoiéticas de individuos sin trastornos clonales hematológicos aparentes(85).
Estas células madre hematopoiéticas mutadas dan lugar a progenies inmunes alteradas que influyen inmunidad del huésped(186). Se reconoce que las mutaciones CHIP más frecuentes promueven la inflamación derivada de mieloides a través de una mayor producción de citoquinas inflamatorias.
Además, CHIP está relacionado con el cáncer no hematológico de varias maneras. CHIP se asocia con un mayor riesgo de desarrollar tumores sólidos, como cáncer de pulmón, cáncer de riñón, linfoma y sarcoma(187).
CHIP se observa en el 20 %-30 % de los pacientes con cáncer en general y está determinado por terapias anticancerígenas(188), y se asocia con un mayor riesgo de muerte a través de la progresión de los tumores primarios no hematológicos(189). Sin embargo, se sabe poco sobre las relaciones entre la disfunción inmune en CHIP y la inmunidad antitumoral durante el inicio, la progresión y la respuesta a las terapias del tumor(190).
Los trabajos futuros que profundizan en la interfaz tumor-inmune dentro del contexto de CHIP están preparados para revelar biomarcadores de pronóstico e intervenciones potencialmente antiinflamatorias para atenuar el inicio y la progresión de la enfermedad.
Microbiomas y cáncer: patogénesis de la enfermedad, respuesta a la terapia y orientación terapéutica
Otra complejidad ambiental que trasciende al huésped per se, además de las discutidas anteriormente, involucra al microbioma humano, compuesto por microbios y sus genomas(191).
Está cada vez más establecido que el microbioma impacta profundamente y de manera variable la patogénesis de las enfermedades y los resultados de la terapia en las neoplasias malignas humanas(192), con microbiomas polimórficos recientemente como una característica fenotípica del cáncer que habilita el sello(4). Cada vez hay más evidencia del impacto de la microbiota intestinal y de otros tejidos en la patogénesis del cáncer, así como en las respuestas a la terapia, junto con implicaciones intrigantes para la microbiota intratumoral. .
El papel de los microbios intestinales en la patogénesis y la respuesta terapéutica.
Los microbios del intestino tienen diversos efectos sobre la inmunidad y el cáncer a través de varios mecanismos diferentes.
Pueden modular directamente los componentes celulares que promueven la carcinogénesis, por ejemplo, en el cáncer colorrectal(193), e influir en la inmunidad sistémica y antitumoral(194).
La importancia de la microbiota intestinal en la configuración de las respuestas al bloqueo de los puntos de control inmunológico se demostró por primera vez en modelos preclínicos(59) y fue seguida rápidamente. por estudios históricos en cohortes humanas(61,62,195), ahora respaldados por una serie de estudios que demuestran asociaciones pronósticas de la microbiota intestinal con cánceres humano(192).
Es importante destacar que estamos ganando más claridad sobre los taxones y las características funcionales de la microbiota intestinal que están asociadas con la respuesta y resistencia a la inmunoterapia, con datos que sugieren que la presencia de taxones desfavorables en el intestino se asocia con inflamación sistémica y deterioro de la inmunidad antitumoral junto con un mayor riesgo de eventos adversos(196).
Estudios recientes también han sugerido que la ingesta dietética puede afectar la microbiota intestinal y la respuesta y toxicidad de la inmunoterapia,(197,198,199), proporcionando así un ángulo manejable para la intervención terapéutica.
El papel de los microbios intratumorales en la patogénesis y la respuesta al tratamiento.
La secuenciación de ácidos nucleicos y los análisis de imágenes han revelado la presencia de microbios intratumorales en varios cánceres humanos(200,201,202,203,204).
La composición y la carga microbiana se han asociado en ciertos tipos de tumores con las respuestas al tratamiento y la supervivencia del paciente(203,205). Los avances en los enfoques multiómicos han proporcionado información mecanicista sobre el papel de los microbios intratumorales dentro del TME oral y colorrectal(204). Inicialmente, los estudios del microbioma intratumoral se centraron en los cánceres gastrointestinales y los sitios de la mucosa accesibles a los microbios.
Sin embargo, en 2020, dos estudios emblemáticos informaron comunidades bacterianas específicas de tipos de tumores en muchos tipos de cáncer sólido(206,207), lo que sugiere un potencial diagnóstico que involucra ácidos nucleicos bacterianos en la sangre de pacientes con cáncer(207).
Un estudio informó que las bacterias intratumorales eran predominantemente intracelulares en -cánceres gastrointestinales(206). De manera similar, se han informado comunidades fúngicas intratumorales específicas del tipo de cáncer(208,209).
Sin embargo, un nuevo análisis de datos reciente ha planteado preocupaciones importantes sobre la prevalencia de microbiomas específicos del tipo de tumor, en particular dentro de las células cancerosas, lo que resalta la contaminación microbiana en conjuntos de datos, contaminación del ADN humano de referencias microbianas y problemas de aprendizaje automático(210,211).
El reanálisis de imágenes bacterianas cuestionó la localización bacteriana dentro de las células epiteliales del cáncer de mama(206), sugiriendo en cambio su presencia en macrófagos y estroma intersticial.
La reproducción de hallazgos sobre hongos intratumorales en cánceres de páncreas también ha enfrentado desafíos(212,213,214) Por lo tanto, si bien existe evidencia convincente de la presencia de microbios que infectan tumores en tipos de cáncer específicos, como el cáncer colorrectal ( 200,201,204,215), ahora existe controversia con respecto a la generalidad de un microbioma genuino específico del tipo de tumor en todos los tipos de cáncer humano.
Se necesita más investigación para determinar si la presencia de comunidades bacterianas y fúngicas intratumorales es exclusiva de casos específicos o es un fenómeno más extendido, y en qué tipos de cáncer son más prevalentes y funcionalmente impactantes.
Dirigirse a los microbios intestinales y tumorales para el tratamiento, la interceptación y la prevención del cáncer
Un creciente conjunto de evidencia respalda la modulación de la microbiota como una estrategia para mejorar los resultados del tratamiento del cáncer(192), incluidos los trasplantes de microbiota fecal(216,217) o la inoculación con especies microbianas bien definidas y guiadas por mecanismos(218). Otras estrategias incluyen la eliminación de especies promotoras de tumores dentro del TME a través de enfoques antibióticos dirigidos(219).
A medida que avancemos, será imperativo desarrollar enfoques estandarizados para evaluar los microbios patógenos (y beneficiosos) que actúan a distancia de los tejidos intestinales y mucosos que habitan naturalmente, así como dentro de los tumores.
Debemos mejorar nuestra comprensión mecanicista de cómo los microbios intestinales e intratumorales dan forma directa e indirectamente a la patogénesis del cáncer y, por lo tanto, impactan los resultados de los pacientes.
Una oportunidad corolaria es aprovechar esta información para identificar biomarcadores microbianos para la prevención o el pronóstico del cáncer, ayudar en el diseño y prueba de estrategias terapéuticas destinadas a modular los microbios y, por lo tanto, tratar mejor el cáncer y, en última instancia, incluso prevenir o interceptar la transformación maligna.
Estrategias futuras destinadas a aprovechar las complejidades del cáncer
Sistemas experimentales para modelar el cáncer y su complejidad.
Los modelos murinos de cáncer humano genéticamente modificados (y los tradicionales trasplantes de células cancerosas) han iluminado durante 40 años los mecanismos de tumorigénesis en varios pasos y la progresión maligna, y cada vez más, las respuestas y la resistencia adaptativa a las terapias, esta última habilitada por la posibilidad de ensamblar factores de edad y enfermedad, cohortes emparejadas para realizar ensayos preclínicos(220,221,222).
Sin embargo, el espectro de complejidades del cáncer discutidas en esta revisión no puede abordarse completamente en dichos modelos, incluida, por ejemplo, la evaluación del impacto de las interacciones gen-ambiente (GxE).
El estudio de grandes cohortes humanas, como el Biobanco del Reino Unido y “All of Us”, nos ha hecho conscientes del impacto significativo de los factores de confusión, incluida la estructura de la población, el origen étnico y la exposición ambiental, en los resultados de los análisis genéticos y terapéuticos. focalización.
Esta sencilla lección, que por su naturaleza es en gran medida observacional, aún no ha sido adoptada de manera efectiva por los experimentadores biomédicos.
Por lo tanto, no es sorprendente que los experimentos en animales consanguíneos, destinados a respaldar los vínculos causales y mecanicistas entre genotipos y fenotipos sugeridos por la genética humana, no puedan traducirse en humanos.
Estos modelos experimentales genéticamente restringidos han suscitado un debate sobre las limitaciones de los modelos animales para la investigación del cáncer y han alimentado la noción de que los seres humanos son el mejor modelo para estudiar enfermedades humanas, a pesar de la realidad de que realizar experimentos para abordar plenamente los mecanismos subyacentes es muy difícil, si no imposible en humanos(223).
De cara al futuro, para abordar los desafíos de las complejidades del cáncer y la traducción de su iluminación a mejores tratamientos para los cánceres humanos, prevemos un mayor refinamiento y utilización de otros sistemas modelo, tanto en modelos de ratones genéticamente diversos como ex vivo.
Modelos que involucran células y tejidos humanos.
En un enfoque para modelar los efectos de GxE en el cáncer, se puede adoptar la diversidad genética del ratón. Dicha diversidad se puede lograr a través de estudios en poblaciones genéticas de referencia (GRP), como BXD y Collaborative Cross GRP(224,225) o en ratones exógamos, como los ratones exógamos de diversidad,(226) que colectivamente capturan aproximadamente el 90% de la variación genética en ratones de laboratorio, superando la diversidad genética presente en los humanos.
Estos ratones genéticamente diversos pueden usarse no sólo para estudiar perturbaciones genéticas que protegen o predisponen al desarrollo del cáncer (por ejemplo, generar animales F1 de un GRP cruzado con una cepa que porta un oncogén), así como para estudiar modificadores ambientales que van desde desafíos dietéticos, factores estresantes, hasta intervenciones farmacológicas(227).
Estos estudios poblacionales longitudinales en ratones tienen la posibilidad de (1) modelar la diversidad genética presente en las poblaciones humanas, (2) estudiar las interacciones GxE, (3) permitir el acceso a tumores y tejidos internos para la caracterización molecular, y (4) mapear los genes responsables de un fenotipo determinado mediante el análisis del locus de rasgos cuantitativos (QTL).
Un desafío técnico no resuelto para el futuro será desarrollar medios eficientes para la ingeniería genética de modelos de cáncer exógenos para producir la notable diversidad de tumores de órganos específicos que portan las mismas mutaciones oncogénicas que los cánceres humanos afines en cohortes susceptibles de ensayos preclínicos.
Ciertamente, el campo del cáncer tiene mucho que ganar al adoptar la diversidad genética para modelar el inicio y la progresión del tumor, y utilizar estos modelos para ensayos preclínicos de nuevos medicamentos contra el cáncer y combinaciones guiadas por mecanismos, avanzando en el conocimiento y su aplicación a la medicina del cáncer.
Más allá de todas las innumerables variaciones de modelos murinos de cáncer humano, prevemos que los sistemas ex vivo que involucran células y tejidos humanos se refinarán cada vez más y se aplicarán de manera creativa para dilucidar los mecanismos subyacentes a la complejidad del cáncer y desarrollar ensayos «de precisión/personalizados» tanto para probar hipótesis terapéuticas como para probar hipótesis terapéuticas. y guiar las decisiones de tratamiento para pacientes con cáncer individuales.
Una vez que se hizo evidente que las células madre tisulares, solas o con células estromales, podían propagarse ex vivo como estructuras similares a tejidos en una matriz 3D,(228,229), los cultivos organoides se han utilizado como puente entre los cultivos 2D simplistas y la complejidad de los cultivos humanos in vivo.
Los organoides ofrecen enfoques para propagar tumores ex vivo en tres dimensiones, representando las características patológicas del tumor parental, expandiendo así el número de sistemas modelo derivados de pacientes susceptibles de experimentación con el objetivo de complementar las observaciones de modelos de ratón.
Los organoides pueden establecerse a partir de células madre específicas de células cancerosas o de tejidos adultos, así como de células madre pluripotentes (inducidas o embrionarias)(230), aunque estas últimas tienen el desafío adicional de recapitular fielmente las señales bioquímicas y la organización espacial de los tejidos/ desarrollo tumoral.
A pesar del desafío de la heterogeneidad intratumoral y el sesgo en el muestreo de tejidos, donde los eventos subclonales pueden no estar bien representados en modelos de tumores propagados ex vivo de pacientes, se ha demostrado que los organoides derivados de pacientes mantienen eventos clonales tumorales(231), y sus firmas transcriptómicas pueden ser similares a las tumores de los que se derivaron(232).
Se han logrado avances recientes en el desarrollo de organoides para reflejar más efectivamente el TME observado en los cánceres humanos, con métodos de cocultivo de organoides de linfocitos/tumores en desarrollo(233), como medios para crear una vasculatura funcional.
Otra limitación del enfoque organoide en el modelado del cáncer es la posible falta de ECM específica del cáncer y del microambiente físico (rigidez, presión del líquido intersticial, vascularización, etc.). Actualmente, la mayoría de los métodos de cultivo de organoides utilizan Matrigel, colágeno o una mezcla de ambos. Aprovechando el campo de la bioingeniería, se están desarrollando muchos materiales sintéticos que permiten la generación de entornos más controlados que permiten la modulación de la rigidez del tejido(234).
Prevemos el desarrollo futuro de «tumoroides» que recapitulen más completamente todas estas características destacadas de la TME auténtica.
Los explantes derivados de tumores y las tecnologías de órganos en chips ofrecen soluciones adicionales a estos desafíos, que pueden mejorar el descubrimiento y desarrollo de fármacos contra el cáncer al permitir modelos humanos de cáncer apropiados y diversos con TME representativos.
Las tecnologías de órgano en chip proporcionan modelos más elaborados de enfermedad, empleando microfluidos para modelar las interfaces tisulares y comenzar a recapitular el complejo TME, lo que permite la investigación de las interacciones tumor/estroma y las pruebas de fármacos anticancerígenos(235,236,237). Los explantes tumorales derivados de pacientes son iniciados cultivando fragmentos o secciones gruesas (vibratomo) de tejidos tumorales frescos, lo que permite el mantenimiento a corto plazo de las características histopatológicas de los tumores originales.
Dado que se puede mantener la morfología espacial del tumor, estos enfoques pueden ofrecer la capacidad de combinar pruebas de fármacos ex vivo con análisis de biomarcadores para identificar predictores de la respuesta al fármaco y descifrar el impacto del TME en la respuesta y la resistencia al fármaco(238).
Los ratones con xenoinjerto derivado de pacientes (PDX) también facilitan las pruebas in vivo de fármacos en el contexto de un tumor humano en un entorno inmunodeficiente; Los ratones PDX parcialmente “humanizados” que presentan algunas características del sistema inmunológico humano han ampliado aún más su uso para evaluar inmunoterapias(239), aunque será necesario un mayor refinamiento para modelar completamente la complejidad y la diversidad polimórfica de la respuesta inmune humana a los tumores humanos en ratones huéspedes.
Además, los modelos PDX son engorrosos, costosos y, en muchos casos, incapaces de replicar la complejidad y heterogeneidad de los tumores humanos y sus microambientes.
Los fragmentos de tumores derivados de pacientes (PDTF) abordan algunos de estos problemas al permitir pruebas ex vivo del impacto de nuevos fármacos en múltiples fragmentos de tumores obtenidos de pacientes(240,241).
La plataforma PDTF permite la perturbación de fragmentos de tumores frescos o congelados conservando el microambiente del original. tumor, aunque sólo brevemente. Fundamentalmente, en un estudio se demostró que la respuesta del PDTF ex vivo a la inhibición del punto de control es altamente concordante con la respuesta clínica del mismo paciente a los agentes anti-proteína de muerte celular programada 1 (anti-PD1), lo que ilustra el valor potencial de esta plataforma(242).
Si bien todavía son limitados en algunos aspectos (como la viabilidad a corto plazo de los fragmentos y la pérdida de células inmunitarias innatas y vasculatura funcional), prevemos que mejoras adicionales en esta plataforma contribuirán significativamente a nuestra comprensión de las respuestas (y la resistencia) al cáncer.
Explotando el otro lado de la adicción a los oncogenes
El estudio de la resistencia a las terapias dirigidas durante las últimas dos décadas ha revelado que la reactivación de la vía de señalización inhibida por fármacos mediante mutaciones secundarias es un mecanismo dominante de resistencia.
Al inhibir las señales a las que la célula cancerosa es adicta, podemos hacer que la célula cancerosa evolucione en la dirección de una señalización superior.
Esto plantea la pregunta de si también podemos dirigir las células cancerosas en la dirección opuesta, es decir, evolucionar hacia un fenotipo menos maligno mediante una señalización oncogénica reducida.
Al considerar este concepto, es importante darse cuenta de que las células cancerosas se esfuerzan por optimizar, en lugar de maximizar, la señalización mitogénica.
En consonancia con esto, la evidencia emergente indica que una mayor estimulación de la señalización mitogénica en las células cancerosas puede ser tan tóxica como la inhibición de estas señales, al activar aún más las vías de respuesta al estrés celular(243).
De hecho, una combinación de hiperactivación de la señalización oncogénica y la inhibición de las señales asociadas a las respuestas al estrés son muy efectivas en modelos animales de cáncer de colon. Sorprendentemente, la resistencia a esta combinación se asoció con una regulación negativa de la señalización oncogénica y la pérdida de la capacidad oncogénica(244).
Una preocupación obvia en este enfoque terapéutico es la presencia de muchas células aparentemente normales en el soma envejecido que albergan mutaciones oncogénicas (consulte la sección de promoción de tumores).
Estas células pueden ser estimuladas para expandirse mediante fármacos que activan aún más la señalización oncogénica. Sin embargo, en modelos animales de adenomas intestinales precancerosos, un aumento adicional en la señalización Wnt por el tratamiento con cloruro de litio se asoció con una reducción en la formación de adenomas en lugar de un aumento, lo que indica que también en un estado precanceroso, hay un escenario en el que una mayor señal oncogénica no es mejor(245). Consistentemente, los estudios epidemiológicos muestran que el tratamiento a largo plazo de pacientes con trastorno bipolar con cloruro de litio se asocia con un riesgo reducido de cáncer colorrectal(246).
Aunque los riesgos y obstáculos de este enfoque paradójico deben ser abordar, dirigir las células cancerosas hacia un fenotipo menos maligno a través de la hiperactivación deliberada de la señalización oncogénica es prometedor para futuras terapias contra el cáncer.
Traducir los descubrimientos a los pacientes: ensayos clínicos de próxima generación
Muchos descubrimientos de la ciencia básica nos informan sobre los primeros pasos de la tumorigénesis. Por lo tanto, existe una oportunidad de desarrollar ensayos clínicos que prueben medicamentos en cánceres en etapa temprana y plataformas de prevención.
Dos tipos distintos de ensayos preoperatorios representan diseños atractivos para el desarrollo de fármacos y su traducción clínica. En primer lugar, los ensayos clínicos neoadyuvantes evalúan la eficacia de fármacos administrados con intención terapéutica varios meses antes de la cirugía. Esos ensayos también permiten la investigación de los mecanismos de resistencia(247).
En segundo lugar, los ensayos de ventana de oportunidad no están diseñados para evaluar la eficacia de los medicamentos, pero son particularmente atractivos para explorar los mecanismos de acción y la farmacodinamia de nuevos medicamentos administrados por un período corto (normalmente unos pocos días) antes del inicio de la terapia contra el cáncer(248).
Además, en un entorno de prevención, la categorización de los estudios como primarios, secundarios y terciarios es un marco atractivo que puede resultar útil para el desarrollo futuro de fármacos si cuenta con el respaldo de sociedades científicas(249).
Los estudios de quimioprevención primaria describen el uso de medicamentos para prevenir el cáncer en personas de alto riesgo.
Se necesitarán definiciones precisas de factores clínicos, genómicos, demográficos y ambientales capaces de resaltar las poblaciones de pacientes en riesgo para permitir dichos estudios.
Esta capacidad podría lograrse mediante estudios epidemiológicos y preclínicos combinados, facilitados mediante la mejora de las capacidades de recopilación de datos clínicos, incluida la patología tumoral facilitada por IA digital(21,250). Se necesitarán nuevos criterios de valoración biológicos que capturen los efectos del tratamiento asociados con el desarrollo deficiente del cáncer como indicadores tempranos del beneficio del tratamiento.
Los estudios de quimioprevención secundaria se refieren a la evaluación de posibles intervenciones en pacientes con condiciones premalignas establecidas.
Existe la necesidad de una evaluación prospectiva y aleatoria de agentes prometedores del espacio preclínico en este entorno terapéutico para múltiples indicaciones de cáncer(251).
Los estudios de quimioprevención terciaria implican enfoques para reducir el riesgo de recaída del cáncer, localmente o en otros órganos.
En esta categoría, el ADN tumoral circulante tendrá un papel cada vez más importante como herramienta para detectar de forma no invasiva la enfermedad residual mínima (ERM) como biomarcador y criterio de valoración temprano de eficacia terapéutica, ya que esta tecnología es capaz de detectar y determinar la respuesta al tratamiento en pacientes con enfermedad micrometastásicos que no es identificable mediante imágenes clínicas(252,253,254,255,256).
Al ser el cáncer una enfermedad impulsada por mecanismos biológicos, surge la necesidad de establecer nuevos sistemas de clasificación basados en la biología de la enfermedad, además de los actuales basados en el órgano de origen. Las clasificaciones multiómicas podrían acelerar drásticamente el desarrollo de fármacos dirigidos a vías biológicas a través de ensayos clínicos independientes de órganos(257).
Las secciones anteriores de esta revisión han demostrado que el cáncer es una enfermedad compleja impulsada por mecanismos biológicos múltiples e interrelacionados.
Por lo tanto, estas complejidades deberían integrarse en retratos que recapitulen la biología completa de cada paciente con cáncer. Se están desarrollando ensayos que prueban la utilidad clínica de estos retratos moleculares completos y múltiples del cáncer(258), aunque en la actualidad validar la utilidad de los componentes individuales de los análisis múltiples en un gran número de pacientes es un desafío.
Además, es casi seguro que el futuro de la terapia contra el cáncer sea uno de combinación de objetivos múltiples con clases distintivas de fármacos contra el cáncer que alteren los mecanismos que impulsan los tumores, así como las manifestaciones sistémicas.
Por lo tanto, a medida que avance el conocimiento sobre los mecanismos moleculares y celulares que impulsan las complejidades del cáncer y sus efectos patogénicos, estos y otros diseños innovadores de ensayos terapéuticos clínicos (y preclínicos) serán fundamentales para traducir los resultados en beneficio de los pacientes con cáncer.
Observaciones finales
Mejorar la cantidad de vida de los pacientes con cáncer sigue siendo un objetivo general de la investigación sobre el cáncer.
De cara al futuro, mejorar la calidad y la duración de la vida (“cáncer sin enfermedad”), disminuir la carga de síntomas y mejorar la sostenibilidad, asequibilidad y accesibilidad de la atención médica enfrentan desafíos monumentales pero, en última instancia, manejables, que son posibles si se adopta y comprende mejor la compleja biología del cáncer, la enfermedad, local y sistémicamente, como se elabora en este documento.
A pesar de los avances en nuestra comprensión de la enfermedad, se estima que la incidencia del cáncer aumentará entre un 20% y un 50% en los próximos 20 años, lo que refleja en parte el envejecimiento de la población, el aumento de la obesidad y los factores ambientales multifacéticos que influyen en la aparición de enfermedades malignas progresión del cáncer, una realidad que exige inversiones continuas y esfuerzos redoblados para abordar sus complejidades.
Además, la incidencia de cánceres de aparición temprana está aumentando(259) por razones que no están claras. Una cuestión general relacionada con la nueva era de abordar las complejidades del cáncer involucra las manifestaciones sistémicas de la enfermedad y el impacto de las exposiciones e influencias ambientales (desde la contaminación críptica y bien reconocida hasta las dietas y la microbiota poco saludables y más) hasta el curso de la tumorigénesis en múltiples etapas, progresión maligna y resistencia a la terapia en todo el espectro de cánceres humanos.
Esclarecer y abordar con éxito estas complejidades se beneficiará de la contratación en la empresa de investigación del cáncer de científicos y académicos clínicos con experiencia en áreas fuera de la oncología, como ciencia de datos, modelos matemáticos, neurociencia, inflamación y autoinmunidad, envejecimiento, metabolismo, endocrinología y músculo. fisiología.
Esta expansión de la experiencia y las capacidades requerirá nuevas oportunidades de financiamiento e investigación que crucen disciplinas y fronteras internacionales y vayan más allá de los mecanismos de financiamiento tradicionales (en gran parte gubernamentales), análogos al Programa CRUK/NCI Cancer Grand Challenges, de base filantrópica, dirigido específicamente a fomentar múltiples -colaboraciones disciplinarias para abordar la gran cantidad de preguntas delineadas en esta revisión.
Además, en un momento en el que los compromisos con la formación académica clínica y las trayectorias profesionales posteriores están amenazados en todo el mundo, existe una necesidad apremiante de revitalizar los programas académicos clínicos de formación de MD/PhD, junto con becas de investigación post-MD en lugar de doctorados formales así como iniciativas innovadoras y mecanismos de financiación para “comprar” tiempo protegido para que los médicos científicos participen sustancialmente en la investigación traslacional.
Es comprensible que se hayan centrado muchos esfuerzos en lograr “curas” para esta enfermedad; si bien es un objetivo estratégico apropiado, la realidad de la adaptación neodarwiniana de las células cancerosas, facilitada por la inestabilidad del genoma y la evolución mutacional, así como por la plasticidad epigenética, sugiere que es posible que todas las formas de cáncer nunca sean completamente curables.
Sin embargo, de manera optimista, dilucidar y abordar los mecanismos que permiten la plasticidad fenotípica y la flexibilidad de las células cancerosas a través de la tumorigénesis, la progresión maligna y la resistencia adaptativa a la terapia puede brindar oportunidades para transformar el cáncer en una enfermedad crónica, en gran medida asintomática, aumentando la supervivencia y mejorando la calidad de la vida de los pacientes con cáncer.
Las conexiones entre el envejecimiento y el cáncer se extienden más allá de la acumulación de mutaciones. Comprender cómo los paisajes de órganos, tejidos y tumores cambian con la edad y con diversas exposiciones ambientales, y cómo esos cambios influyen en todas las etapas de la evolución del cáncer, desde la selección de clones iniciados hasta la metástasis, será esencial para ganar más batallas y tal vez incluso guerras contra el cáncer.
Durante las próximas dos décadas, mientras el mundo se enfrenta a un envejecimiento de la población en el que el cáncer afectará al menos a uno de cada tres y la incidencia mundial aumentará, este estudio puede ayudar a proporcionar un marco lógico para las iniciativas destinadas a abordar las nubes de complejidad aquí descrita, con el objetivo de prevenir cada vez más la aparición del cáncer, reducir la incidencia del cáncer sintomático, así como diagnosticar los cánceres más tempranamente, aliviando colectivamente el sufrimiento y prolongando la calidad y cantidad de vida de quienes padecen esta desalentadora enfermedad.
EPÍLOGO
La realidad de la adaptación neodarwiniana de las células cancerosas, facilitada por la inestabilidad del genoma y la evolución mutacional, así como por la plasticidad epigenética, sugiere que es posible que todas las formas de cáncer nunca sean completamente curables.
Esto en cierta manera recuerda lo dicho en una fábula con cierto grado de humor negro que dice asi: Se dice que JESÚS estaba en un lugar cercano a la antigua Roma, donde se congregaron cientos de personas enfermas que buscaban que JESÚS los curara con un milagro. Así pues, vino a JESÚS un ciego y lo hizo ver, vino un paralítico y lo hizo andar , vino uno con artritis y lo curó y así sucesivamente, hasta que vino una persona con una bola muy visible en el cuello, JESÚS la revisa y le dice, lo siento mucho, ud tiene Cáncer.
Referencias Bibliográficas
- 1
S. Wishart
Is Cancer a Genetic Disease or a Metabolic Disease?EBiomedicine, 2 (2015), pp. 478-479
View PDFView articleView in ScopusGoogle Scholar - 2
Hanahan, R.A. Weinberg
The hallmarks of cancerCell, 100 (2000), pp. 57-70
View PDFView articleView in ScopusGoogle Scholar - 3
Hanahan, R.A. Weinberg
Hallmarks of cancer: the next generationCell, 144 (2011), pp. 646-674
View PDFView articleView in ScopusGoogle Scholar - 4
Hanahan
Hallmarks of Cancer: New DimensionsCancer Discov., 12 (2022), pp. 31-46
View at publisher CrossRefView in ScopusGoogle Scholar - 5
Hanahan, M. Monje
Cancer hallmarks intersect with neuroscience in the tumor microenvironmentCancer Cell, 41 (2023), pp. 573-580
View PDFView articleView in ScopusGoogle Scholar - 6
Brennan, G. Davey-Smith
Identifying Novel Causes of Cancers to Enhance Cancer Prevention: New Strategies Are NeededJ. Natl. Cancer Inst., 114 (2022), pp. 353-360
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 7
Clocchiatti, E. Cora, Y. Zhang, G.P. Dotto
Sexual dimorphism in cancerNat. Rev. Cancer, 16 (2016), pp. 330-339
View at publisher CrossRefView in ScopusGoogle Scholar - 8
Haupt, F. Caramia, S.L. Klein, J.B. Rubin, Y. Haupt
Sex disparities matter in cancer development and therapyNat. Rev. Cancer, 21 (2021), pp. 393-407
View at publisher CrossRefView in ScopusGoogle Scholar - 9
Li, Z. Lan, W. Liao, J.W. Horner, X. Xu, J. Liu, Y. Yoshihama, S. Jiang, H.S. Shim, M. Slotnik, et al.
Histone demethylase KDM5D upregulation drives sex differences in colon cancerNature, 619 (2023), pp. 632-639
Google Scholar - 10
C. Özdemir, G.P. Dotto
Sex Hormones and Anticancer ImmunityClin. Cancer Res., 25 (2019), pp. 4603-4610
View at publisher CrossRefView in ScopusGoogle Scholar - 11
P. Vellano, M.G. White, M.C. Andrews, M. Chelvanambi, R.G. Witt, J.R. Daniele, M. Titus, J.L. McQuade, F. Conforti, E.M. Burton, et al.
Androgen receptor blockade promotes response to BRAF/MEK-targeted therapyNature, 606 (2022), pp. 797-803
View at publisher CrossRefView in ScopusGoogle Scholar - 12
Martincorena, P.J. Campbell
Somatic mutation in cancer and normal cellsScience, 349 (2015), pp. 1483-1489
View at publisher CrossRefView in ScopusGoogle Scholar - 13
C. Fowler, P.H. Jones
Somatic Mutation: What Shapes the Mutational Landscape of Normal Epithelia?Cancer Discov., 12 (2022), pp. 1642-1655
View at publisher CrossRefView in ScopusGoogle Scholar - 14
Berenblum, P. Shubik
The role of croton oil applications, associated with a single painting of a carcinogen, in tumour induction of the mouse’s skinBr. J. Cancer, 1 (1947), pp. 379-382
View at publisher CrossRefView in ScopusGoogle Scholar - 15
F. Friedewald, P. Rous
THE INITIATING AND PROMOTING ELEMENTS IN TUMOR PRODUCTION: AN ANALYSIS OF THE EFFECTS OF TAR, BENZPYRENE, AND METHYLCHOLANTHRENE ON RABBIT SKINJ. Exp. Med., 80 (1944), pp. 101-126
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 16
Riva, A.R. Pandiri, Y.R. Li, A. Droop, J. Hewinson, M.A. Quail, V. Iyer, R. Shepherd, R.A. Herbert, P.J. Campbell, et al.
The mutational signature profile of known and suspected human carcinogens in miceNat. Genet., 52 (2020), pp. 1189-1197
View at publisher CrossRefView in ScopusGoogle Scholar - 17
Guerra, A.J. Schuhmacher, M. Cañamero, P.J. Grippo, L. Verdaguer, L. Pérez-Gallego, P. Dubus, E.P. Sandgren, M. Barbacid
Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult miceCancer Cell, 11 (2007), pp. 291-302
View PDFView articleView in ScopusGoogle Scholar - 18
Bailleul, M.A. Surani, S. White, S.C. Barton, K. Brown, M. Blessing, J. Jorcano, A. Balmain
Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoterCell, 62 (1990), pp. 697-708
View PDFView articleView in ScopusGoogle Scholar - 19
Alonso-Curbelo, Y.J. Ho, C. Burdziak, J.L.V. Maag, J.P. Morris, R. Chandwani, H.A. Chen, K.M. Tsanov, F.M. Barriga, W. Luan, et al.
A gene-environment-induced epigenetic program initiates tumorigenesisNature, 590 (2021), pp. 642-648
View at publisher CrossRefView in ScopusGoogle Scholar - 20
N. Wong, G. Ramsingh, A.L. Young, C.A. Miller, W. Touma, J.S. Welch, T.L. Lamprecht, D. Shen, J. Hundal, R.S. Fulton, et al.
Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemiaNature, 518 (2015), pp. 552-555
View at publisher CrossRefView in ScopusGoogle Scholar - 21
Hill, E.L. Lim, C.E. Weeden, C. Lee, M. Augustine, K. Chen, F.C. Kuan, F. Marongiu, E.J. Evans, D.A. Moore, et al.
Lung adenocarcinoma promotion by air pollutantsNature, 616 (2023), pp. 159-167
View at publisher CrossRefView in ScopusGoogle Scholar - 22
Kadariya, C.W. Menges, J. Talarchek, K.Q. Cai, A.J. Klein-Szanto, R.A. Pietrofesa, M. Christofidou-Solomidou, M. Cheung, B.T. Mossman, A. Shukla, et al.
Inflammation-Related IL1β/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant MesotheliomaCancer Prev. Res. (Phila), 9 (2016), pp. 406-414
View in ScopusGoogle Scholar - 23
Moody, S. Senkin, S.M.A. Islam, J. Wang, D. Nasrollahzadeh, R. Cortez Cardoso Penha, S. Fitzgerald, E.N. Bergstrom, J. Atkins, Y. He, et al.
Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidenceNat. Genet., 53 (2021), pp. 1553-1563
View at publisher CrossRefView in ScopusGoogle Scholar - 24
B. Lowenfels, P. Maisonneuve, G. Cavallini, R.W. Ammann, P.G. Lankisch, J.R. Andersen, E.P. Dimagno, A. Andrén-Sandberg, L. Domellöf
Pancreatitis and the risk of pancreatic cancer. International pancreatitis study groupN. Engl. J. Med., 328 (1993), pp. 1433-1437
View in ScopusGoogle Scholar - 25
H. Collins Jr., M. Feldman, J.S. Fordtran
Colon cancer, dysplasia, and surveillance in patients with ulcerative colitis. A critical reviewN. Engl. J. Med., 316 (1987), pp. 1654-1658
View in ScopusGoogle Scholar - 26
George, S. Prasad, Z. Mahmood, Y. Shukla
Studies on glyphosate-induced carcinogenicity in mouse skin: a proteomic approachJ. Proteomics, 73 (2010), pp. 951-964
View PDFView articleView in ScopusGoogle Scholar - 27
Luo, H. Ge
Hot Tea Consumption and Esophageal Cancer Risk: A Meta-Analysis of Observational StudiesFront. Nutr., 9 (2022), p. 831567
View in ScopusGoogle Scholar - 28
Naik, S.B. Larsen, N.C. Gomez, K. Alaverdyan, A. Sendoel, S. Yuan, L. Polak, A. Kulukian, S. Chai, E. Fuchs
Inflammatory memory sensitizes skin epithelial stem cells to tissue damageNature, 550 (2017), pp. 475-480
View at publisher CrossRefView in ScopusGoogle Scholar - 29
B. Alexandrov, J. Kim, N.J. Haradhvala, M.N. Huang, A.W. Tian Ng, Y. Wu, A. Boot, K.R. Covington, D.A. Gordenin, E.N. Bergstrom, et al.
The repertoire of mutational signatures in human cancerNature, 578 (2020), pp. 94-101
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 30
Kanarek, B. Petrova, D.M. Sabatini
Dietary modifications for enhanced cancer therapyNature, 579 (2020), pp. 507-517
View at publisher CrossRefView in ScopusGoogle Scholar - 31
Bejarano, M.J.C. Jordāo, J.A. Joyce
Therapeutic Targeting of the Tumor MicroenvironmentCancer Discov., 11 (2021), pp. 933-959
View at publisher CrossRefView in ScopusGoogle Scholar - 32
Plaks, N. Kong, Z. Werb
The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells?Cell Stem Cell, 16 (2015), pp. 225-238
View PDFView articleView in ScopusGoogle Scholar - 33
Debaugnies, S. Rodríguez-Acebes, J. Blondeau, M.A. Parent, M. Zocco, Y. Song, V. de Maertelaer, V. Moers, M. Latil, C. Dubois, et al.
RHOJ controls EMT-associated resistance to chemotherapyNature, 616 (2023), pp. 168-175
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 34
E. de Visser, J.A. Joyce
The evolving tumor microenvironment: From cancer initiation to metastatic outgrowthCancer Cell, 41 (2023), pp. 374-403
View PDFView articleView in ScopusGoogle Scholar - 35
Cañellas-Socias, C. Cortina, X. Hernando-Momblona, S. Palomo-Ponce, E.J. Mulholland, G. Turon, L. Mateo, S. Conti, O. Roman, M. Sevillano, et al.
Metastatic recurrence in colorectal cancer arises from residual EMP1+ cellsNature, 611 (2022), pp. 603-613
View at publisher CrossRefView in ScopusGoogle Scholar - 36
S. Kim, Y. Gao, T. Welte, H. Wang, J. Liu, M. Janghorban, K. Sheng, Y. Niu, A. Goldstein, N. Zhao, et al.
Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanismsNat. Cell Biol., 21 (2019), pp. 1113-1126
View at publisher CrossRefView in ScopusGoogle Scholar - 37
Lavie, A. Ben-Shmuel, N. Erez, R. Scherz-Shouval
Cancer-associated fibroblasts in the single-cell eraNat. Cancer, 3 (2022), pp. 793-807
View at publisher CrossRefView in ScopusGoogle Scholar - 38
Dai, P.J. Cimino, K.H. Gouin 3rd, C.A. Grzelak, A. Barrett, A.R. Lim, A. Long, S. Weaver, L.T. Saldin, A. Uzamere, et al.
Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brainNat. Cancer, 3 (2022), pp. 25-42
View in ScopusGoogle Scholar - 39
S. Di Martino, A.R. Nobre, C. Mondal, I. Taha, E.F. Farias, E.J. Fertig, A. Naba, J.A. Aguirre-Ghiso, J.J. Bravo-Cordero
A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancyNat. Cancer, 3 (2022), pp. 90-107
View in ScopusGoogle Scholar - 40
E. Fane, Y. Chhabra, G.M. Alicea, D.A. Maranto, S.M. Douglass, M.R. Webster, V.W. Rebecca, G.E. Marino, F. Almeida, B.L. Ecker, et al.
Stromal changes in the aged lung induce an emergence from melanoma dormancyNature, 606 (2022), pp. 396-405
View at publisher CrossRefView in ScopusGoogle Scholar - 41
Ombrato, E. Nolan, I. Kurelac, A. Mavousian, V.L. Bridgeman, I. Heinze, P. Chakravarty, S. Horswell, E. Gonzalez-Gualda, G. Matacchione, et al.
Metastatic-niche labelling reveals parenchymal cells with stem featuresNature, 572 (2019), pp. 603-608
View at publisher CrossRefView in ScopusGoogle Scholar - 42
Klemm, R.R. Maas, R.L. Bowman, M. Kornete, K. Soukup, S. Nassiri, J.P. Brouland, C.A. Iacobuzio-Donahue, C. Brennan, V. Tabar, et al.
Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune CellsCell, 181 (2020), pp. 1643-1660.e17
View PDFView articleView in ScopusGoogle Scholar - 43
Á.F. Álvarez-Prado, R.R. Maas, K. Soukup, F. Klemm, M. Kornete, F.S. Krebs, V. Zoete, S. Berezowska, J.P. Brouland, A.F. Hottinger, et al.
Immunogenomic analysis of human brain metastases reveals diverse immune landscapes across genetically distinct tumorsCell Rep. Med., 4 (2023), p. 100900
View PDFView articleView in ScopusGoogle Scholar - 44
Risom, D.R. Glass, I. Averbukh, C.C. Liu, A. Baranski, A. Kagel, E.F. McCaffrey, N.F. Greenwald, B. Rivero-Gutiérrez, S.H. Strand, et al.
Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stromaCell, 185 (2022), pp. 299-310.e18
View PDFView articleView in ScopusGoogle Scholar - 45
W. Jackson, J.R. Fischer, V.R.T. Zanotelli, H.R. Ali, R. Mechera, S.D. Soysal, H. Moch, S. Muenst, Z. Varga, W.P. Weber, et al.
The single-cell pathology landscape of breast cancerNature, 578 (2020), pp. 615-620
View at publisher CrossRefView in ScopusGoogle Scholar - 46
M. Schürch, S.S. Bhate, G.L. Barlow, D.J. Phillips, L. Noti, I. Zlobec, P. Chu, S. Black, J. Demeter, D.R. McIlwain, et al.
Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive FrontCell, 183 (2020), p. 838
View PDFView articleView in ScopusGoogle Scholar - 47
J. Sammut, M. Crispin-Ortuzar, S.F. Chin, E. Provenzano, H.A. Bardwell, W. Ma, W. Cope, A. Dariush, S.J. Dawson, J.E. Abraham, et al.
Multi-omic machine learning predictor of breast cancer therapy responseNature, 601 (2022), pp. 623-629
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 48
Liu, Y. Zhao, P. Kong, Y. Liu, J. Huang, E. Xu, W. Wei, G. Li, X. Cheng, L. Xue, et al.
Integrated multi-omics profiling yields a clinically relevant molecular classification for esophageal squamous cell carcinomaCancer Cell, 41 (2023), pp. 181-195.e9
View PDFView articleCrossRefGoogle Scholar - 49
Croci, R. Santalla Méndez, S. Temme, K. Soukup, N. Fournier, A. Zomer, R. Colotti, V. Wischnewski, U. Flögel, R.B. van Heeswijk, et al.
Multispectral fluorine-19 MRI enables longitudinal and noninvasive monitoring of tumor-associated macrophagesSci. Transl. Med., 14 (2022), p. eabo2952
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 50
Dmitrieva-Posocco, A. Dzutsev, D.F. Posocco, V. Hou, W. Yuan, V. Thovarai, I.A. Mufazalov, M. Gunzer, I.P. Shilovskiy, M.R. Khaitov, et al.
Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal CancerImmunity, 50 (2019), pp. 166-180.e7
View PDFView articleView in ScopusGoogle Scholar - 51
Dou, K. Ghosh, M.G. Vizioli, J. Zhu, P. Sen, K.J. Wangensteen, J. Simithy, Y. Lan, Y. Lin, Z. Zhou, et al.
Cytoplasmic chromatin triggers inflammation in senescence and cancerNature, 550 (2017), pp. 402-406
View at publisher CrossRefGoogle Scholar - 52
Milanovic, D.N.Y. Fan, D. Belenki, J.H.M. Däbritz, Z. Zhao, Y. Yu, J.R. Dörr, L. Dimitrova, D. Lenze, I.A. Monteiro Barbosa, et al.
Senescence-associated reprogramming promotes cancer stemnessNature, 553 (2018), pp. 96-100
View at publisher CrossRefView in ScopusGoogle Scholar - 53
Cai, F. Aguilera, B. Ramdas, S.V. Daulatabad, R. Srivastava, J.J. Kotzin, M. Carroll, G. Wertheim, A. Williams, S.C. Janga, et al.
Targeting Bim via a lncRNA Morrbid Regulates the Survival of Preleukemic and Leukemic CellsCell Rep., 31 (2020), p. 107816
View PDFView articleView in ScopusGoogle Scholar - 54
Biavasco, E. Lettera, K. Giannetti, D. Gilioli, S. Beretta, A. Conti, S. Scala, D. Cesana, P. Gallina, M. Norelli, et al.
Oncogene-induced senescence in hematopoietic progenitors features myeloid restricted hematopoiesis, chronic inflammation and histiocytosisNat. Commun., 12 (2021), p. 4559
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 55
Tu, G. Bhagat, G. Cui, S. Takaishi, E.A. Kurt-Jones, B. Rickman, K.S. Betz, M. Penz-Oesterreicher, O. Bjorkdahl, J.G. Fox, et al.
Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in miceCancer Cell, 14 (2008), pp. 408-419
View PDFView articleView in ScopusGoogle Scholar - 56
Deng, H. Liang, M. Xu, X. Yang, B. Burnette, A. Arina, X.D. Li, H. Mauceri, M. Beckett, T. Darga, et al.
STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic TumorsImmunity, 41 (2014), pp. 843-852
View PDFView articleView in ScopusGoogle Scholar - 57
R. Woo, M.B. Fuertes, L. Corrales, S. Spranger, M.J. Furdyna, M.Y. Leung, R. Duggan, Y. Wang, G.N. Barber, K.A. Fitzgerald, et al.
STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumorsImmunity, 41 (2014), pp. 830-842
View PDFView articleView in ScopusGoogle Scholar - 58
Ahn, T. Xia, A. Rabasa Capote, D. Betancourt, G.N. Barber
Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying CellsCancer Cell, 33 (2018), pp. 862-873.e5
View PDFView articleView in ScopusGoogle Scholar - 59
Vétizou, J.M. Pitt, R. Daillère, P. Lepage, N. Waldschmitt, C. Flament, S. Rusakiewicz, B. Routy, M.P. Roberti, C.P. Duong, et al.
Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiotaScience, 350 (2015), pp. 1079-1084
View at publisher CrossRefView in ScopusGoogle Scholar - 60
Sivan, L. Corrales, N. Hubert, J.B. Williams, K. Aquino-Michaels, Z.M. Earley, F.W. Benyamin, Y.M. Lei, B. Jabri, M.L. Alegre, et al.
Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacyScience, 350 (2015), pp. 1084-1089
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 61
Gopalakrishnan, C.N. Spencer, L. Nezi, A. Reuben, M.C. Andrews, T.V. Karpinets, P.A. Prieto, D. Vicente, K. Hoffman, S.C. Wei, et al.
Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patientsScience, 359 (2018), pp. 97-103
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 62
Matson, J. Fessler, R. Bao, T. Chongsuwat, Y. Zha, M.L. Alegre, J.J. Luke, T.F. Gajewski
The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patientsScience, 359 (2018), pp. 104-108
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 63
Xing, M. Wang, A.A. Ajibade, P. Tan, C. Fu, L. Chen, M. Zhu, Z.Z. Hao, J. Chu, X. Yu, et al.
Microbiota regulate innate immune signaling and protective immunity against cancerCell Host Microbe, 29 (2021), pp. 959-974.e7
View PDFView articleView in ScopusGoogle Scholar - 64
-C.J. Tsay, B.G. Wu, I. Sulaiman, K. Gershner, R. Schluger, Y. Li, T.A. Yie, P. Meyn, E. Olsen, L. Perez, et al.
Lower Airway Dysbiosis Affects Lung Cancer ProgressionCancer Discov., 11 (2021), pp. 293-307
View at publisher CrossRefView in ScopusGoogle Scholar - 65
Villemin, A. Six, B.A. Neville, T.D. Lawley, M.J. Robinson, G. Bakdash
The heightened importance of the microbiome in cancer immunotherapyTrends Immunol., 44 (2023), pp. 44-59
View PDFView articleView in ScopusGoogle Scholar - 66
Zhao, D. Teng, L. Yang, X. Xu, J. Chen, T. Jiang, A.Y. Feng, Y. Zhang, D.T. Frederick, L. Gu, et al.
Myeloid-derived itaconate suppresses cytotoxic CD8+ T cells and promotes tumour growthNat. Metab., 4 (2022), pp. 1660-1673
View at publisher CrossRefView in ScopusGoogle Scholar - 67
Low, Z. Li, J. Blenis
Metabolite activation of tumorigenic signaling pathways in the tumor microenvironmentSci. Signal., 15 (2022), p. eabj4220
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 68
Juarez, D.A. Fruman
Targeting the Mevalonate Pathway in CancerTrends Cancer, 7 (2021), pp. 525-540
View PDFView articleView in ScopusGoogle Scholar - 69
J. Chen, G.N. Li, X.J. Li, L.X. Wei, M.J. Fu, Z.L. Cheng, Z. Yang, G.Q. Zhu, X.D. Wang, C. Zhang, et al.
Targeting IRG1 reverses the immunosuppressive function of tumor-associated macrophages and enhances cancer immunotherapySci. Adv., 9 (2023), p. eadg0654
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 70
N. Messmer, C.S. Netherby, D. Banik, S.I. Abrams
Tumor-induced myeloid dysfunction and its implications for cancer immunotherapyCancer Immunol. Immunother., 64 (2015), pp. 1-13
View at publisher CrossRefGoogle Scholar - 71
I. Gabrilovich, S. Nagaraj
Myeloid-derived suppressor cells as regulators of the immune systemNat. Rev. Immunol., 9 (2009), pp. 162-174
View at publisher CrossRefView in ScopusGoogle Scholar - 72
M. Kortlever, N.M. Sodir, C.H. Wilson, D.L. Burkhart, L. Pellegrinet, L. Brown Swigart, T.D. Littlewood, G.I. Evan
Myc Cooperates with Ras by Programming Inflammation and Immune SuppressionCell, 171 (2017), pp. 1301-1315.e14
View PDFView articleView in ScopusGoogle Scholar - 73
Rahib, M.R. Wehner, L.M. Matrisian, K.T. Nead
Estimated Projection of US Cancer Incidence and Death to 2040JAMA Netw. Open, 4 (2021), Article e214708
View at publisher
This article is free to access.CrossRefGoogle Scholar - 74
Fane, A.T. Weeraratna
How the ageing microenvironment influences tumour progressionNat. Rev. Cancer, 20 (2020), pp. 89-106
View at publisher CrossRefView in ScopusGoogle Scholar - 75
I. Rozhok, J. DeGregori
Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutationsProc. Natl. Acad. Sci. USA, 112 (2015), pp. 8914-8921
View at publisher CrossRefView in ScopusGoogle Scholar - 76
Chhabra, A.T. Weeraratna
Fibroblasts in cancer: unity in heterogeneityCell, 186 (2023), pp. 1580-1609
View PDFView articleView in ScopusGoogle Scholar - 77
H.J. Hoeijmakers
DNA damage, aging, and cancerN. Engl. J. Med., 361 (2009), pp. 1475-1485
View in ScopusGoogle Scholar - 78
Marongiu, J. DeGregori
The sculpting of somatic mutational landscapes by evolutionary forces and their impacts on aging-related diseaseMol. Oncol., 16 (2022), pp. 3238-3258
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 79
Ahmad, N. Jahn, S. Jaiswal
Clonal Hematopoiesis and Its Impact on Human HealthAnnu. Rev. Med., 74 (2023), pp. 249-260
View at publisher CrossRefView in ScopusGoogle Scholar - 80
Olafsson, R.E. McIntyre, T. Coorens, T. Butler, H. Jung, P.S. Robinson, H. Lee-Six, M.A. Sanders, K. Arestang, C. Dawson, et al.
Somatic Evolution in Non-neoplastic IBD-Affected ColonCell, 182 (2020), pp. 672-684.e11
View PDFView articleView in ScopusGoogle Scholar - 81
Nanki, M. Fujii, M. Shimokawa, M. Matano, S. Nishikori, S. Date, A. Takano, K. Toshimitsu, Y. Ohta, S. Takahashi, et al.
Somatic inflammatory gene mutations in human ulcerative colitis epitheliumNature, 577 (2020), pp. 254-259
View at publisher CrossRefView in ScopusGoogle Scholar - 82
Kakiuchi, K. Yoshida, M. Uchino, T. Kihara, K. Akaki, Y. Inoue, K. Kawada, S. Nagayama, A. Yokoyama, S. Yamamoto, et al.
Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitisNature, 577 (2020), pp. 260-265
View at publisher CrossRefView in ScopusGoogle Scholar - 83
C. Fowler, C. King, C. Bryant, M.W.J. Hall, R. Sood, S.H. Ong, E. Earp, D. Fernandez-Antoran, J. Koeppel, S.C. Dentro, et al.
Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body SiteCancer Discov., 11 (2021), pp. 340-361
View at publisher CrossRefView in ScopusGoogle Scholar - 84
Yokoyama, N. Kakiuchi, T. Yoshizato, Y. Nannya, H. Suzuki, Y. Takeuchi, Y. Shiozawa, Y. Sato, K. Aoki, S.K. Kim, et al.
Age-related remodelling of oesophageal epithelia by mutated cancer driversNature, 565 (2019), pp. 312-317
View at publisher CrossRefView in ScopusGoogle Scholar - 85
Jaiswal, B.L. Ebert
Clonal hematopoiesis in human aging and diseaseScience, 366 (2019), p. eaan4673
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 86
Kerr, C. Anderson, S.M. Lippman
Physical activity, sedentary behaviour, diet, and cancer: an update and emerging new evidenceLancet Oncol., 18 (2017), pp. e457-e471
View PDFView articleView in ScopusGoogle Scholar - 87
Furman, J. Campisi, E. Verdin, P. Carrera-Bastos, S. Targ, C. Franceschi, L. Ferrucci, D.W. Gilroy, A. Fasano, G.W. Miller, et al.
Chronic inflammation in the etiology of disease across the life spanNat. Med., 25 (2019), pp. 1822-1832
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 88
K. Turrell, R. Orha, N.J. Guppy, A. Gillespie, M. Guelbert, C. Starling, S. Haider, C.M. Isacke
Age-associated microenvironmental changes highlight the role of PDGF-C in ER+ breast cancer metastatic relapseNat. Cancer, 4 (2023), pp. 468-484
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 89
L. Kiemen, A.M. Braxton, M.P. Grahn, K.S. Han, J.M. Babu, R. Reichel, A.C. Jiang, B. Kim, J. Hsu, F. Amoa, et al.
CODA: quantitative 3D reconstruction of large tissues at cellular resolutionNat. Methods, 19 (2022), pp. 1490-1499
View at publisher CrossRefView in ScopusGoogle Scholar - 90
Berry, N.A. Giraldo, B.F. Green, T.R. Cottrell, J.E. Stein, E.L. Engle, H. Xu, A. Ogurtsova, C. Roberts, D. Wang, et al.
Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockadeScience, 372 (2021), p. eaba2609
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 91
Drapela, A.P. Gomes
The aging lung reawakens dormant tumor cellsNat. Cancer, 4 (2023), pp. 442-443
View at publisher CrossRefView in ScopusGoogle Scholar - 92
Wang, L. Lankhorst, R. Bernards
Exploiting senescence for the treatment of cancerNat. Rev. Cancer, 22 (2022), pp. 340-355
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 93
Jochems, B. Thijssen, G. De Conti, R. Jansen, Z. Pogacar, K. Groot, L. Wang, A. Schepers, C. Wang, H. Jin, et al.
The Cancer SENESCopedia: A delineation of cancer cell senescenceCell Rep., 36 (2021), p. 109441
View PDFView articleView in ScopusGoogle Scholar - 94
Demaria, M.N. O’Leary, J. Chang, L. Shao, S. Liu, F. Alimirah, K. Koenig, C. Le, N. Mitin, A.M. Deal, et al.
Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer RelapseCancer Discov., 7 (2017), pp. 165-176
View in ScopusGoogle Scholar - 95
J. Baker, T. Wijshake, T. Tchkonia, N.K. LeBrasseur, B.G. Childs, B. van de Sluis, J.L. Kirkland, J.M. van Deursen
Clearance of p16Ink4a-positive senescent cells delays ageing-associated disordersNature, 479 (2011), pp. 232-236
View at publisher CrossRefView in ScopusGoogle Scholar - 96
J. Baker, B.G. Childs, M. Durik, M.E. Wijers, C.J. Sieben, J. Zhong, R.A. Saltness, K.B. Jeganathan, G.C. Verzosa, A. Pezeshki, et al.
Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespanNature, 530 (2016), pp. 184-189
View at publisher CrossRefView in ScopusGoogle Scholar - 97
Wang, H. Jin, F. Jochems, S. Wang, C. Lieftink, I.M. Martinez, G. De Conti, F. Edwards, R.L. de Oliveira, A. Schepers, et al.
cFLIP suppression and DR5 activation sensitize senescent cancer cells to senolysisNat. Cancer, 3 (2022), pp. 1284-1299
View at publisher CrossRefView in ScopusGoogle Scholar - 98
Ruscetti, J.P. Morris 4th, R. Mezzadra, J. Russell, J. Leibold, P.B. Romesser, J. Simon, A. Kulick, Y.-J. Ho, M. Fennell, et al.
Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas CancerCell, 181 (2020), pp. 424-441.e21
View PDFView articleView in ScopusGoogle Scholar - 99
Kang, Q. Xu, T.D. Martin, M.Z. Li, M. Demaria, L. Aron, T. Lu, B.A. Yankner, J. Campisi, S.J. Elledge
The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4Science, 349 (2015), p. aaa5612
Google Scholar - 100
Li, M.C. Simon
Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor MicroenvironmentsDev. Cell, 54 (2020), pp. 183-195
View PDFView articleView in ScopusGoogle Scholar - 101
C. Kimmelman, E. White
Autophagy and Tumor MetabolismCell Metab., 25 (2017), pp. 1037-1043
View PDFView articleView in ScopusGoogle Scholar - 102
Al-Zoughbi, W. Al-Zhoughbi, J. Huang, G.S. Paramasivan, H. Till, M. Pichler, B. Guertl-Lackner, G. Hoefler
Tumor macroenvironment and metabolismSemin. Oncol., 41 (2014), pp. 281-295
Google Scholar - 103
Li, Y. Yang, B. Zhang, X. Lin, X. Fu, Y. An, Y. Zou, J.X. Wang, Z. Wang, T. Yu
Lactate metabolism in human health and diseaseSignal Transduct. Target. Ther., 7 (2022), p. 305
View at publisher CrossRefGoogle Scholar - 104
Hui, J.M. Ghergurovich, R.J. Morscher, C. Jang, X. Teng, W. Lu, L.A. Esparza, T. Reya, Le Zhan, J. Yanxiang Guo, et al.
Glucose feeds the TCA cycle via circulating lactateNature, 551 (2017), pp. 115-118
View at publisher CrossRefView in ScopusGoogle Scholar - 105
Faubert, K.Y. Li, L. Cai, C.T. Hensley, J. Kim, L.G. Zacharias, C. Yang, Q.N. Do, S. Doucette, D. Burguete, et al.
Lactate Metabolism in Human Lung TumorsCell, 171 (2017), pp. 358-371.e9
View PDFView articleView in ScopusGoogle Scholar - 106
Du, H. Zhang, Y. Feng, D. Zhan, S. Li, C. Tu, J. Liu, J. Wang
Tumour-derived small extracellular vesicles contribute to the tumour progression through reshaping the systemic immune macroenvironmentBr. J. Cancer, 128 (2023), pp. 1249-1266
View at publisher CrossRefView in ScopusGoogle Scholar - 107
Goldman, L.N. Adler, E. Hajaj, T. Croese, N. Darzi, S. Galai, H. Tishler, Y. Ariav, D. Lavie, L. Fellus-Alyagor, et al.
Early Infiltration of Innate Immune Cells to the Liver Depletes HNF4α and Promotes Extrahepatic CarcinogenesisCancer Discov., 13 (2023), pp. 1616-1635
View at publisher CrossRefView in ScopusGoogle Scholar - 108
G. Burfeind, X. Zhu, M.A. Norgard, P.R. Levasseur, C. Huisman, A.C. Buenafe, B. Olson, K.A. Michaelis, E.R. Torres, S. Jeng, et al.
Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancereLife, 9 (2020), p. e54095
Google Scholar - 109
M. Argilés, F.J. López-Soriano, B. Stemmler, S. Busquets
Cancer-associated cachexia – understanding the tumour macroenvironment and microenvironment to improve managementNat. Rev. Clin. Oncol., 20 (2023), pp. 250-264
View at publisher CrossRefView in ScopusGoogle Scholar - 110
R. Pryce, D.J. Wang, T.A. Zimmers, M.C. Ostrowski, D.C. Guttridge
Cancer cachexia: involvement of an expanding macroenvironmentCancer Cell, 41 (2023), pp. 581-584
View PDFView articleView in ScopusGoogle Scholar - 111
Martin, L. Birdsell, N. Macdonald, T. Reiman, M.T. Clandinin, L.J. McCargar, R. Murphy, S. Ghosh, M.B. Sawyer, V.E. Baracos
Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass indexJ. Clin. Oncol., 31 (2013), pp. 1539-1547
View in ScopusGoogle Scholar - 112
E. Baracos, L. Martin, M. Korc, D.C. Guttridge, K.C.H. Fearon
Cancer-associated cachexiaNat. Rev. Dis. Primers, 4 (2018), p. 17105
View in ScopusGoogle Scholar - 113
Ferrer, T.G. Anthony, J.S. Ayres, G. Biffi, J.C. Brown, B.J. Caan, E.M. Cespedes Feliciano, A.P. Coll, R.F. Dunne, M.D. Goncalves, et al.
Cachexia: A systemic consequence of progressive, unresolved diseaseCell, 186 (2023), pp. 1824-1845
View PDFView articleView in ScopusGoogle Scholar - 114
Neyroud, O. Laitano, A. Dasgupta, C. Lopez, R.E. Schmitt, J.Z. Schneider, D.W. Hammers, H.L. Sweeney, G.A. Walter, J. Doles, et al.
Blocking muscle wasting via deletion of the muscle-specific E3 ligase MuRF1 impedes pancreatic tumor growthCommun. Biol., 6 (2023), p. 519
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 115
Ternes, M. Tsenkova, V.I. Pozdeev, M. Meyers, E. Koncina, S. Atatri, M. Schmitz, J. Karta, M. Schmoetten, A. Heinken, et al.
The gut microbial metabolite formate exacerbates colorectal cancer progressionNat. Metab., 4 (2022), pp. 458-475
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 116
Pati, W. Irfan, A. Jameel, S. Ahmed, R.K. Shahid
Obesity and Cancer: A Current Overview of Epidemiology, Pathogenesis, Outcomes, and ManagementCancers, 15 (2023), p. 485
View at publisher CrossRefView in ScopusGoogle Scholar - 117
H. Schmitz, A.M. Campbell, M.M. Stuiver, B.M. Pinto, A.L. Schwartz, G.S. Morris, J.A. Ligibel, A. Cheville, D.A. Galvão, C.M. Alfano, et al.
Exercise is medicine in oncology: engaging clinicians to help patients move through cancerCA Cancer J. Clin., 69 (2019), pp. 468-484
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 118
McTiernan
Mechanisms linking physical activity with cancerNat. Rev. Cancer, 8 (2008), pp. 205-211
View at publisher CrossRefView in ScopusGoogle Scholar - 119
M. Moore, S. Lee, T. Olsen, M. Morselli, A.R. Strumwasser, A.J. Lin, Z. Zhou, A. Abrishami, S.M. Garcia, J. Bribiesca, et al.
Conserved multi-tissue transcriptomic adaptations to exercise training in humans and miceCell Rep., 42 (2023), p. 112499
View PDFView articleView in ScopusGoogle Scholar - 120
Wei, N.M. Riley, X. Lyu, X. Shen, J. Guo, S.H. Raun, M. Zhao, M.D. Moya-Garzon, H. Basu, A. Sheng-Hwa Tung, et al.
Organism-wide, cell-type-specific secretome mapping of exercise training in miceCell Metab., 35 (2023), pp. 1261-1279.e11
View PDFView articleCrossRefView in ScopusGoogle Scholar - 121
V. Liberti, Z. Dai, S.E. Wardell, J.A. Baccile, X. Liu, X. Gao, R. Baldi, M. Mehrmohamadi, M.O. Johnson, N.S. Madhukar, et al.
A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural ProductCell Metab., 26 (2017), pp. 648-659.e8
View PDFView articleView in ScopusGoogle Scholar - 122
Altea-Manzano, A.M. Cuadros, L.A. Broadfield, S.M. Fendt
Nutrient metabolism and cancer in the in vivo context: a metabolic game of give and takeEMBO Rep., 21 (2020), Article e50635
View in ScopusGoogle Scholar - 123
Tajan, K.H. Vousden
Dietary Approaches to Cancer TherapyCancer Cell, 37 (2020), pp. 767-785
View PDFView articleView in ScopusGoogle Scholar - 124
W. Locasale
Diet and Exercise in Cancer MetabolismCancer Discov., 12 (2022), pp. 2249-2257
View at publisher CrossRefView in ScopusGoogle Scholar - 125
Di Biase, C. Lee, S. Brandhorst, B. Manes, R. Buono, C.W. Cheng, M. Cacciottolo, A. Martin-Montalvo, R. de Cabo, M. Wei, et al.
Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor CytotoxicityCancer Cell, 30 (2016), pp. 136-146
View PDFView articleView in ScopusGoogle Scholar - 126
Valdemarin, I. Caffa, A. Persia, A.L. Cremonini, L. Ferrando, L. Tagliafico, A. Tagliafico, A. Guijarro, F. Carbone, S. Ministrini, et al.
Safety and Feasibility of Fasting-Mimicking Diet and Effects on Nutritional Status and Circulating Metabolic and Inflammatory Factors in Cancer Patients Undergoing Active TreatmentCancers, 13 (2021), p. 4013
View at publisher CrossRefView in ScopusGoogle Scholar - 127
D.K. Maddocks, D. Athineos, E.C. Cheung, P. Lee, T. Zhang, N.J.F. van den Broek, G.M. Mackay, C.F. Labuschagne, D. Gay, F. Kruiswijk, et al.
Modulating the therapeutic response of tumours to dietary serine and glycine starvationNature, 544 (2017), pp. 372-376
View at publisher CrossRefView in ScopusGoogle Scholar - 128
Gao, S.M. Sanderson, Z. Dai, M.A. Reid, D.E. Cooper, M. Lu, J.P. Richie, A. Ciccarella, A. Calcagnotto, P.G. Mikhael, et al.
Dietary methionine influences therapy in mouse cancer models and alters human metabolismNature, 572 (2019), pp. 397-401
View in ScopusGoogle Scholar - 129
C. Lien, A.M. Westermark, Y. Zhang, C. Yuan, Z. Li, A.N. Lau, K.M. Sapp, B.M. Wolpin, M.G. Vander Heiden
Low glycaemic diets alter lipid metabolism to influence tumour growthNature, 599 (2021), pp. 302-307
View at publisher CrossRefView in ScopusGoogle Scholar - 130
D. Hopkins, C. Pauli, X. Du, D.G. Wang, X. Li, D. Wu, S.C. Amadiume, M.D. Goncalves, C. Hodakoski, M.R. Lundquist, et al.
Suppression of insulin feedback enhances the efficacy of PI3K inhibitorsNature, 560 (2018), pp. 499-503
View at publisher CrossRefView in ScopusGoogle Scholar - 131
R. Taylor, J.N. Falcone, L.C. Cantley, M.D. Goncalves
Developing dietary interventions as therapy for cancerNat. Rev. Cancer, 22 (2022), pp. 452-466
View at publisher CrossRefView in ScopusGoogle Scholar - 132
Pariollaud, K.A. Lamia
Cancer in the Fourth Dimension: What Is the Impact of Circadian Disruption?Cancer Discov., 10 (2020), pp. 1455-1464
View at publisher CrossRefView in ScopusGoogle Scholar - 133
Zhang, N.F. Lahens, H.I. Ballance, M.E. Hughes, J.B. Hogenesch
A circadian gene expression atlas in mammals: implications for biology and medicineProc. Natl. Acad. Sci. USA, 111 (2014), pp. 16219-16224
View at publisher CrossRefView in ScopusGoogle Scholar - 134
Gotoh, M. Vila-Caballer, C.S. Santos, J. Liu, J. Yang, C.V. Finkielstein
The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cellsMol. Biol. Cell, 25 (2014), pp. 3081-3093
View in ScopusGoogle Scholar - 135
B. Chan, G.C.G. Parico, J.L. Fribourgh, L.H. Ibrahim, M.J. Bollong, C.L. Partch, K.A. Lamia
CRY2 missense mutations suppress P53 and enhance cell growthProc. Natl. Acad. Sci. USA, 118 (2021), Article e2101416118
View in ScopusGoogle Scholar - 136
L. Huber, S.J. Papp, A.B. Chan, E. Henriksson, S.D. Jordan, A. Kriebs, M. Nguyen, M. Wallace, Z. Li, C.M. Metallo, et al.
CRY2 and FBXL3 Cooperatively Degrade c-MYCMol. Cell, 64 (2016), pp. 774-789
View PDFView articleView in ScopusGoogle Scholar - 137
Liu, C.P. Selby, Y. Yang, L.A. Lindsey-Boltz, X. Cao, K. Eynullazada, A. Sancar
Circadian regulation of c-MYC in miceProc. Natl. Acad. Sci. USA, 117 (2020), pp. 21609-21617
View at publisher CrossRefView in ScopusGoogle Scholar - 138
Pariollaud, L.H. Ibrahim, E. Irizarry, R.M. Mello, A.B. Chan, B.J. Altman, R.J. Shaw, M.J. Bollong, R.L. Wiseman, K.A. Lamia
Circadian disruption enhances HSF1 signaling and tumorigenesis in Kras-driven lung cancerSci. Adv., 8 (2022), p. eabo1123
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 139
Wang, C. Barnoud, M. Cenerenti, M. Sun, I. Caffa, B. Kizil, R. Bill, Y. Liu, R. Pick, L. Garnier, et al.
Dendritic cells direct circadian anti-tumour immune responsesNature, 614 (2023), pp. 136-143
Google Scholar - 140
K. Chun, B.M. Fortin, R.C. Fellows, A.N. Habowski, A. Verlande, W.A. Song, A.L. Mahieu, A.E.Y.T. Lefebvre, J.N. Sterrenberg, L.M. Velez, et al.
Disruption of the circadian clock drives Apc loss of heterozygosity to accelerate colorectal cancerSci. Adv., 8 (2022), p. eabo2389
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 141
Qu, G. Zhang, H. Qu, A. Vu, R. Wu, H. Tsukamoto, Z. Jia, W. Huang, H.J. Lenz, J.N. Rich, et al.
Circadian regulator BMAL1::CLOCK promotes cell proliferation in hepatocellular carcinoma by controlling apoptosis and cell cycleProc. Natl. Acad. Sci. USA, 120 (2023), Article e2214829120
View in ScopusGoogle Scholar - 142
Papagiannakopoulos, M.R. Bauer, S.M. Davidson, M. Heimann, L. Subbaraj, A. Bhutkar, J. Bartlebaugh, M.G. Vander Heiden, T. Jacks
Circadian Rhythm Disruption Promotes Lung TumorigenesisCell Metab., 24 (2016), pp. 324-331
View PDFView articleView in ScopusGoogle Scholar - 143
J. Alasady, M.L. Mendillo
The Multifaceted Role of HSF1 in TumorigenesisAdv. Exp. Med. Biol., 1243 (2020), pp. 69-85
View at publisher CrossRefView in ScopusGoogle Scholar - 144
Diamantopoulou, F. Castro-Giner, F.D. Schwab, C. Foerster, M. Saini, S. Budinjas, K. Strittmatter, I. Krol, B. Seifert, V. Heinzelmann-Schwarz, et al.
The metastatic spread of breast cancer accelerates during sleepNature, 607 (2022), pp. 156-162
View at publisher CrossRefView in ScopusGoogle Scholar - 145
Verlande, S.K. Chun, M.O. Goodson, B.M. Fortin, H. Bae, C. Jang, S. Masri
Glucagon regulates the stability of REV-ERBα to modulate hepatic glucose production in a model of lung cancer-associated cachexiaSci. Adv., 7 (2021), p. eabf3885
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 146
F. Innominato, A. Ballesta, Q. Huang, C. Focan, P. Chollet, A. Karaboué, S. Giacchetti, M. Bouchahda, R. Adam, C. Garufi, et al.
Sex-dependent least toxic timing of irinotecan combined with chronomodulated chemotherapy for metastatic colorectal cancer: Randomized multicenter EORTC 05011 trialCancer Med., 9 (2020), pp. 4148-4159
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 147
C. Qian, T. Kleber, B. Brammer, K.M. Xu, J.M. Switchenko, J.R. Janopaul-Naylor, J. Zhong, M.L. Yushak, R.D. Harvey, C.M. Paulos, et al.
Effect of immunotherapy time-of-day infusion on overall survival among patients with advanced melanoma in the USA (MEMOIR): a propensity score-matched analysis of a single-centre, longitudinal studyLancet Oncol., 22 (2021), pp. 1777-1786
View PDFView articleView in ScopusGoogle Scholar - 148
Winkler, H.S. Venkatesh, M. Amit, T. Batchelor, I.E. Demir, B. Deneen, D.H. Gutmann, S. Hervey-Jumper, T. Kuner, D. Mabbott, et al.
Cancer neuroscience: state of the field, emerging directionsCell, 186 (2023), pp. 1689-1707
View PDFView articleView in ScopusGoogle Scholar - 149
Osswald, E. Jung, F. Sahm, G. Solecki, V. Venkataramani, J. Blaes, S. Weil, H. Horstmann, B. Wiestler, M. Syed, et al.
Brain tumour cells interconnect to a functional and resistant networkNature, 528 (2015), pp. 93-98
View at publisher CrossRefView in ScopusGoogle Scholar - 150
Venkataramani, D.I. Tanev, C. Strahle, A. Studier-Fischer, L. Fankhauser, T. Kessler, C. Körber, M. Kardorff, M. Ratliff, R. Xie, et al.
Glutamatergic synaptic input to glioma cells drives brain tumour progressionNature, 573 (2019), pp. 532-538
View at publisher CrossRefView in ScopusGoogle Scholar - 151
Hausmann, D.C. Hoffmann, V. Venkataramani, E. Jung, S. Horschitz, S.K. Tetzlaff, A. Jabali, L. Hai, T. Kessler, D.D. Azoŕin, et al.
Autonomous rhythmic activity in glioma networks drives brain tumour growthNature, 613 (2023), pp. 179-186
View at publisher CrossRefView in ScopusGoogle Scholar - 152
Venkataramani, Y. Yang, M.C. Schubert, E. Reyhan, S.K. Tetzlaff, N. Wißmann, M. Botz, S.J. Soyka, C.A. Beretta, R.L. Pramatarov, et al.
Glioblastoma hijacks neuronal mechanisms for brain invasionCell, 185 (2022), pp. 2899-2917.e31
View PDFView articleView in ScopusGoogle Scholar - 153
Krishna, A. Choudhury, M.B. Keough, K. Seo, L. Ni, S. Kakaizada, A. Lee, A. Aabedi, G. Popova, B. Lipkin, et al.
Glioblastoma remodelling of human neural circuits decreases survivalNature, 617 (2023), pp. 599-607
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 154
Haroun, J.N. Wood, S. Sikandar
Mechanisms of cancer painFront. Pain Res. (Lausanne), 3 (2022), p. 1030899
View in ScopusGoogle Scholar - 155
Zhang, C. Xie, Y. Lu
Local Anesthetic Lidocaine and Cancer: Insight Into Tumor Progression and RecurrenceFront. Oncol., 11 (2021), p. 669746
View in ScopusGoogle Scholar - 156
Mullard
Vertex’s Nav1.8 inhibitor passes phase II pain pointNat. Rev. Drug Discov., 21 (2022), p. 327
Google Scholar - 157
Heuer, I. Burghaus, M. Gose, T. Kessler, F. Sahm, P. Vollmuth, V. Venkataramani, D. Hoffmann, M. Schlesner, M. Ratliff, et al.
PerSurge (NOA-30) phase II trial of perampanel treatment around surgery in patients with progressive glioblastomaBMC Cancer, 24 (2024), p. 135, 10.1186/s12885-024-11846-1
PMID: 38279087; PMCID: PMC10811925
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 158
L. Britt, J. Cuzick, K.A. Phillips
Key steps for effective breast cancer preventionNat. Rev. Cancer, 20 (2020), pp. 417-436
View at publisher CrossRefView in ScopusGoogle Scholar - 159
Collaborative Group on Hormonal Factors in Breast Cancer
Breast cancer and breastfeeding: collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50302 women with breast cancer and 96973 women without the diseaseLancet, 360 (2002), pp. 187-195
Google Scholar - 160
Islami, Y. Liu, A. Jemal, J. Zhou, E. Weiderpass, G. Colditz, P. Boffetta, M. Weiss
Breastfeeding and breast cancer risk by receptor status–a systematic review and meta-analysisAnn. Oncol., 26 (2015), pp. 2398-2407
View PDFView articleView in ScopusGoogle Scholar - 161
Loi, D. Drubay, S. Adams, G. Pruneri, P.A. Francis, M. Lacroix-Triki, H. Joensuu, M.V. Dieci, S. Badve, S. Demaria, et al.
Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast CancersJ. Clin. Oncol., 37 (2019), pp. 559-569
View at publisher CrossRefView in ScopusGoogle Scholar - 162
Savas, B. Virassamy, C. Ye, A. Salim, C.P. Mintoff, F. Caramia, R. Salgado, D.J. Byrne, Z.L. Teo, S. Dushyanthen, et al.
Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosisNat. Med., 24 (2018), pp. 986-993
View at publisher CrossRefView in ScopusGoogle Scholar - 163
Virassamy, F. Caramia, P. Savas, S. Sant, J. Wang, S.N. Christo, A. Byrne, K. Clarke, E. Brown, Z.L. Teo, et al.
Intratumoral CD8+ T cells with a tissue-resident memory phenotype mediate local immunity and immune checkpoint responses in breast cancerCancer Cell, 41 (2023), pp. 585-601.e8
View PDFView articleView in ScopusGoogle Scholar - 164
Pal, Y. Chen, F. Vaillant, B.D. Capaldo, R. Joyce, X. Song, V.L. Bryant, J.S. Penington, L. Di Stefano, N. Tubau Ribera, et al.
A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breastEMBO J., 40 (2021), Article e107333
View in ScopusGoogle Scholar - 165
Asztalos, P.H. Gann, M.K. Hayes, L. Nonn, C.A. Beam, Y. Dai, E.L. Wiley, D.A. Tonetti
Gene expression patterns in the human breast after pregnancyCancer Prev. Res. (Phila), 3 (2010), pp. 301-311
View in ScopusGoogle Scholar - 166
Koren, J.K. Goodrich, T.C. Cullender, A. Spor, K. Laitinen, H.K. Bäckhed, A. Gonzalez, J.J. Werner, L.T. Angenent, R. Knight, et al.
Host remodeling of the gut microbiome and metabolic changes during pregnancyCell, 150 (2012), pp. 470-480 - 167
M. Hunt, J.A. Foster, L.J. Forney, U.M. Schütte, D.L. Beck, Z. Abdo, L.K. Fox, J.E. Williams, M.K. McGuire, M.A. McGuire
Characterization of the diversity and temporal stability of bacterial communities in human milkPLoS One, 6 (2011), Article e21313 - 168
Cabrera-Rubio, M.C. Collado, K. Laitinen, S. Salminen, E. Isolauri, A. Mira
The human milk microbiome changes over lactation and is shaped by maternal weight and mode of deliveryAm. J. Clin. Nutr., 96 (2012), pp. 544-551 - 169
Camacho-Morales, M. Caba, M. García-Juárez, M.D. Caba-Flores, R. Viveros-Contreras, C. Martínez-Valenzuela
Breastfeeding Contributes to Physiological Immune Programming in the NewbornFront. Pediatr., 9 (2021), p. 744104 - 170
A. Khorana, C.W. Francis, E. Culakova, N.M. Kuderer, G.H. Lyman
Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapyJ. Thromb. Haemost., 5 (2007), pp. 632-634 - 171
A. Khorana, C.W. Francis, E. Culakova, R.I. Fisher, N.M. Kuderer, G.H. Lyman
Thromboembolism in hospitalized neutropenic cancer patientsJ. Clin. Oncol., 24 (2006), pp. 484-490 - 172
I. Mulder, E. Horváth-Puhó, N. van Es, H.W.M. van Laarhoven, L. Pedersen, F. Moik, C. Ay, H.R. Büller, H.T. Sørensen
Venous thromboembolism in cancer patients: a population-based cohort studyBlood, 137 (2021), pp. 1959-1969 - 173
Engelmann, S. Massberg
Thrombosis as an intravascular effector of innate immunityNat. Rev. Immunol., 13 (2013), pp. 34-45 - 174
A. Fuchs, A. Brill, D. Duerschmied, D. Schatzberg, M. Monestier, D.D. Myers, S.K. Wrobleski, T.W. Wakefield, J.H. Hartwig, D.D. Wagner
Extracellular DNA traps promote thrombosisProc. Natl. Acad. Sci. USA, 107 (2010), pp. 15880-15885 - 175
S. Kasthuri, M.B. Taubman, N. Mackman
Role of tissue factor in cancerJ. Clin. Oncol., 27 (2009), pp. 4834-4838 - 176
Anvari, E. Osei, N. Maftoon
Interactions of platelets with circulating tumor cells contribute to cancer metastasisSci. Rep., 11 (2021), p. 15477 - 177
Tawil, R. Bassawon, B. Meehan, A. Nehme, L. Montermini, T. Gayden, N. De Jay, C. Spinelli, S. Chennakrishnaiah, D. Choi, et al.
Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesiclesBlood Adv., 5 (2021), pp. 1682-1694 - 178
Xue, Q. Zhang, Q. Cao, R. Kong, X. Xiang, H. Liu, M. Feng, F. Wang, J. Cheng, Z. Li, et al.
Liver tumour immune microenvironment subtypes and neutrophil heterogeneityNature, 612 (2022), pp. 141-147 - 179
Ballesteros, A. Rubio-Ponce, M. Genua, E. Lusito, I. Kwok, G. Fernández-Calvo, T.E. Khoyratty, E. van Grinsven, S. González-Hernández, J.Á. Nicolás-Ávila, et al.
Co-option of Neutrophil Fates by Tissue EnvironmentsCell, 183 (2020), pp. 1282-1297.e18 - 180
R. Maas, K. Soukup, N. Fournier, M. Massara, S. Galland, M. Kornete, V. Wischnewski, J. Lourenco, D. Croci, Á.F. Álvarez-Prado, et al.
The local microenvironment drives activation of neutrophils in human brain tumorsCell, 186 (2023), pp. 4546-4566.e27 - 181
Ponzetta, R. Carriero, S. Carnevale, M. Barbagallo, M. Molgora, C. Perucchini, E. Magrini, F. Gianni, P. Kunderfranco, N. Polentarutti, et al.
Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human TumorsCell, 178 (2019), pp. 346-360.e24 - 182
Cui, K. Chakraborty, X.A. Tang, G. Zhou, K.Q. Schoenfelt, K.M. Becker, A. Hoffman, Y.F. Chang, A. Blank, C.A. Reardon, et al.
Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesisCell, 184 (2021), pp. 3163-3177.e21 - 183
M. Adrover, S.A.C. McDowell, X.Y. He, D.F. Quail, M. Egeblad
NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular trapsCancer Cell, 41 (2023), pp. 505-526 - 184
A. Khorana, N.M. Kuderer, E. Culakova, G.H. Lyman, C.W. Francis
Development and validation of a predictive model for chemotherapy-associated thrombosisBlood, 111 (2008), pp. 4902-4907 - 185
Wolach, R.S. Sellar, K. Martinod, D. Cherpokova, M. McConkey, R.J. Chappell, A.J. Silver, D. Adams, C.A. Castellano, R.K. Schneider, et al.
Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasmsSci. Transl. Med., 10 (2018), p. eaan8292 - 186
Belizaire, W.J. Wong, M.L. Robinette, B.L. Ebert
Clonal haematopoiesis and dysregulation of the immune systemNat. Rev. Immunol., 23 (2023), pp. 595-610 - 187
P. Kar, P.M. Quiros, M. Gu, T. Jiang, J. Mitchell, R. Langdon, V. Iyer, C. Barcena, M.S. Vijayabaskar, M.A. Fabre, et al.
Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesisNat. Genet., 54 (2022), pp. 1155-1166 - 188
L. Bolton, R.N. Ptashkin, T. Gao, L. Braunstein, S.M. Devlin, D. Kelly, M. Patel, A. Berthon, A. Syed, M. Yabe, et al.
Cancer therapy shapes the fitness landscape of clonal hematopoiesisNat. Genet., 52 (2020), pp. 1219-1226 - 189
C. Coombs, A. Zehir, S.M. Devlin, A. Kishtagari, A. Syed, P. Jonsson, D.M. Hyman, D.B. Solit, M.E. Robson, J. Baselga, et al.
Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical OutcomesCell Stem Cell, 21 (2017), pp. 374-382.e4 - 190
Lee, J. Li, J. Li, S. Fang, J. Zhang, A.T.T. Vo, W. Han, H. Zeng, S. Isgandarova, M. Martinez-Moczygemba, et al.
Tet2 Inactivation Enhances the Antitumor Activity of Tumor-Infiltrating LymphocytesCancer Res., 81 (2021), pp. 1965-1976 - 191
F. Budden, S.D. Shukla, S.F. Rehman, K.L. Bowerman, S. Keely, P. Hugenholtz, D.P.H. Armstrong-James, I.M. Adcock, S.H. Chotirmall, K.F. Chung, et al.
Functional effects of the microbiota in chronic respiratory diseaseLancet Respir. Med., 7 (2019), pp. 907-920 - 192
D. Sepich-Poore, L. Zitvogel, R. Straussman, J. Hasty, J.A. Wargo, R. Knight
The microbiome and human cancerScience, 371 (2021), p. eabc4552 - 193
H. Wong, J. Yu
Gut microbiota in colorectal cancer: mechanisms of action and clinical applicationsNat. Rev. Gastroenterol. Hepatol., 16 (2019), pp. 690-704 - 194
N. Baruch, J. Wang, J.A. Wargo
Gut Microbiota and Antitumor Immunity: Potential Mechanisms for Clinical EffectCancer Immunol. Res., 9 (2021), pp. 365-370 - 195
Routy, E. Le Chatelier, L. Derosa, C.P.M. Duong, M.T. Alou, R. Daillère, A. Fluckiger, M. Messaoudene, C. Rauber, M.P. Roberti, et al.
Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumorsScience, 359 (2018), pp. 91-97 - 196
A. McCulloch, D. Davar, R.R. Rodrigues, J.H. Badger, J.R. Fang, A.M. Cole, A.K. Balaji, M. Vetizou, S.M. Prescott, M.R. Fernandes, et al.
Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1Nat. Med., 28 (2022), pp. 545-556 - 197
N. Spencer, J.L. McQuade, V. Gopalakrishnan, J.A. McCulloch, M. Vetizou, A.P. Cogdill, M.A.W. Khan, X. Zhang, M.G. White, C.B. Peterson, et al.
Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy responseScience, 374 (2021), pp. 1632-1640 - 198
C. Lam, R.E. Araya, A. Huang, Q. Chen, M. Di Modica, R.R. Rodrigues, A. Lopès, S.B. Johnson, B. Schwarz, E. Bohrnsen, et al.
Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironmentCell, 184 (2021), pp. 5338-5356.e21 - 199
C. Simpson, E.R. Shanahan, M. Batten, I.L.M. Reijers, M. Read, I.P. Silva, J.M. Versluis, R. Ribeiro, A.S. Angelatos, J. Tan, et al.
Diet-driven microbial ecology underpins associations between cancer immunotherapy outcomes and the gut microbiomeNat. Med., 28 (2022), pp. 2344-2352 - 200
D. Kostic, D. Gevers, C.S. Pedamallu, M. Michaud, F. Duke, A.M. Earl, A.I. Ojesina, J. Jung, A.J. Bass, J. Tabernero, et al.
Genomic analysis identifies association of Fusobacterium with colorectal carcinomaGenome Res., 22 (2012), pp. 292-298 - 201
Bullman, C.S. Pedamallu, E. Sicinska, T.E. Clancy, X. Zhang, D. Cai, D. Neuberg, K. Huang, F. Guevara, T. Nelson, et al.
Analysis of Fusobacterium persistence and antibiotic response in colorectal cancerScience, 358 (2017), pp. 1443-1448 - 202
T. Geller, M. Barzily-Rokni, T. Danino, O.H. Jonas, N. Shental, D. Nejman, N. Gavert, Y. Zwang, Z.A. Cooper, K. Shee, et al.
Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabineScience, 357 (2017), pp. 1156-1160
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 203
Riquelme, Y. Zhang, L. Zhang, M. Montiel, M. Zoltan, W. Dong, P. Quesada, I. Sahin, V. Chandra, A. San Lucas, et al.
Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer OutcomesCell, 178 (2019), pp. 795-806.e12
View PDFView articleView in ScopusGoogle Scholar - 204
L. Galeano Niño, H. Wu, K.D. LaCourse, A.G. Kempchinsky, A. Baryiames, B. Barber, N. Futran, J. Houlton, C. Sather, E. Sicinska, et al.
Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancerNature, 611 (2022), pp. 810-817
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 205
Serna, F. Ruiz-Pace, J. Hernando, L. Alonso, R. Fasani, S. Landolfi, R. Comas, J. Jimenez, E. Elez, S. Bullman, et al.
Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancerAnn. Oncol., 31 (2020), pp. 1366-1375
View PDFView articleView in ScopusGoogle Scholar - 206
Nejman, I. Livyatan, G. Fuks, N. Gavert, Y. Zwang, L.T. Geller, A. Rotter-Maskowitz, R. Weiser, G. Mallel, E. Gigi, et al.
The human tumor microbiome is composed of tumor type-specific intracellular bacteriaScience, 368 (2020), pp. 973-980
View at publisher
This article is free to access.CrossRefGoogle Scholar - 207
D. Poore, E. Kopylova, Q. Zhu, C. Carpenter, S. Fraraccio, S. Wandro, T. Kosciolek, S. Janssen, J. Metcalf, S.J. Song, et al.
Microbiome analyses of blood and tissues suggest cancer diagnostic approachNature, 579 (2020), pp. 567-574
View at publisher CrossRefView in ScopusGoogle Scholar - 208
Narunsky-Haziza, G.D. Sepich-Poore, I. Livyatan, O. Asraf, C. Martino, D. Nejman, N. Gavert, J.E. Stajich, G. Amit, A. González, et al.
Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactionsCell, 185 (2022), pp. 3789-3806.e17
View PDFView articleView in ScopusGoogle Scholar - 209
B. Dohlman, J. Klug, M. Mesko, I.H. Gao, S.M. Lipkin, X. Shen, I.D. Iliev
A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumorsCell, 185 (2022), pp. 3807-3822.e12
View PDFView articleView in ScopusGoogle Scholar - 210
Gihawi, Y. Ge, J. Lu, D. Puiu, A. Xu, C.S. Cooper, D.S. Brewer, M. Pertea, S.L. Salzberg
Major data analysis errors invalidate cancer microbiome findingsmBio, 14 (2023), Article e0160723
Google Scholar - 211
Gihawi, C.S. Cooper, D.S. Brewer
Caution regarding the specificities of pan-cancer microbial structureMicrob. Genom., 9 (2023), Article mgen001088
Google Scholar - 212
Aykut, S. Pushalkar, R. Chen, Q. Li, R. Abengozar, J.I. Kim, S.A. Shadaloey, D. Wu, P. Preiss, N. Verma, et al.
The fungal mycobiome promotes pancreatic oncogenesis via activation of MBLNature, 574 (2019), pp. 264-267
View at publisher CrossRefView in ScopusGoogle Scholar - 213
A. Fletcher, M.S. Kelly, A.M. Eckhoff, P.J. Allen
Revisiting the intrinsic mycobiome in pancreatic cancerNature, 620 (2023), pp. E1-E6
View at publisher CrossRefView in ScopusGoogle Scholar - 214
Xu, D. Saxena, S. Pushalkar, G. Miller
Reply to: Revisiting the intrinsic mycobiome in pancreatic cancerNature, 620 (2023), pp. E7-E9
View at publisher CrossRefView in ScopusGoogle Scholar - 215
M. Dejea, P. Fathi, J.M. Craig, A. Boleij, R. Taddese, A.L. Geis, X. Wu, C.E. DeStefano Shields, E.M. Hechenbleikner, D.L. Huso, et al.
Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteriaScience, 359 (2018), pp. 592-597
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 216
N. Baruch, I. Youngster, G. Ben-Betzalel, R. Ortenberg, A. Lahat, L. Katz, K. Adler, D. Dick-Necula, S. Raskin, N. Bloch, et al.
Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patientsScience, 371 (2021), pp. 602-609
View at publisher CrossRefView in ScopusGoogle Scholar - 217
Davar, A.K. Dzutsev, J.A. McCulloch, R.R. Rodrigues, J.M. Chauvin, R.M. Morrison, R.N. Deblasio, C. Menna, Q. Ding, O. Pagliano, et al.
Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patientsScience, 371 (2021), pp. 595-602
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 218
Dizman, L. Meza, P. Bergerot, M. Alcantara, T. Dorff, Y. Lyou, P. Frankel, Y. Cui, V. Mira, M. Llamas, et al.
Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trialNat. Med., 28 (2022), pp. 704-712
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 219
A. Holt
Oncomicrobial vaccines: The potential for a Fusobacterium nucleatum vaccine to improve colorectal cancer outcomesCell Host Microbe, 31 (2023), pp. 141-145
View PDFView articleView in ScopusGoogle Scholar - 220
Maertens, M.E. McCurrach, B.S. Braun, T. De Raedt, I. Epstein, T.Q. Huang, J.O. Lauchle, H. Lee, J. Wu, T.P. Cripe, et al.
A Collaborative Model for Accelerating the Discovery and Translation of Cancer TherapiesCancer Res., 77 (2017), pp. 5706-5711
View in ScopusGoogle Scholar - 221
Olson, Y. Li, Y. Lin, E.T. Liu, A. Patnaik
Mouse Models for Cancer Immunotherapy ResearchCancer Discov., 8 (2018), pp. 1358-1365
View at publisher CrossRefView in ScopusGoogle Scholar - 222
J. Drapkin, C.M. Rudin
Advances in Small-Cell Lung Cancer (SCLC) Translational ResearchCold Spring Harb. Perspect. Med., 11 (2021), Article a038240
View at publisher CrossRefView in ScopusGoogle Scholar - 223
H. Nadeau, J. Auwerx
The virtuous cycle of human genetics and mouse models in drug discoveryNat. Rev. Drug Discov., 18 (2019), pp. 255-272
View at publisher CrossRefView in ScopusGoogle Scholar - 224
A. Churchill, D.C. Airey, H. Allayee, J.M. Angel, A.D. Attie, J. Beatty, W.D. Beavis, J.K. Belknap, B. Bennett, W. Berrettini, et al.
The Collaborative Cross, a community resource for the genetic analysis of complex traitsNat. Genet., 36 (2004), pp. 1133-1137
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 225
G. Ashbrook, D. Arends, P. Prins, M.K. Mulligan, S. Roy, E.G. Williams, C.M. Lutz, A. Valenzuela, C.J. Bohl, J.F. Ingels, et al.
A platform for experimental precision medicine: The extended BXD mouse familyCell Syst., 12 (2021), pp. 235-247.e9
View PDFView articleView in ScopusGoogle Scholar - 226
A. Churchill, D.M. Gatti, S.C. Munger, K.L. Svenson
The Diversity Outbred mouse populationMamm. Genome, 23 (2012), pp. 713-718
View at publisher CrossRefView in ScopusGoogle Scholar - 227
D. Halliwill, D.A. Quigley, H.C. Kang, R. Del Rosario, D. Ginzinger, A. Balmain
Panx3 links body mass index and tumorigenesis in a genetically heterogeneous mouse model of carcinogen-induced cancerGenome Med., 8 (2016), p. 83
View at publisher
This article is free to access.View in ScopusGoogle Scholar - 228
Ootani, X. Li, E. Sangiorgi, Q.T. Ho, H. Ueno, S. Toda, H. Sugihara, K. Fujimoto, I.L. Weissman, M.R. Capecchi, et al.
Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell nicheNat. Med., 15 (2009), pp. 701-706
View at publisher CrossRefView in ScopusGoogle Scholar - 229
Pastuła, M. Middelhoff, A. Brandtner, M. Tobiasch, B. Höhl, A.H. Nuber, I.E. Demir, S. Neupert, P. Kollmann, G. Mazzuoli-Weber, et al.
Three-Dimensional Gastrointestinal Organoid Culture in Combination with Nerves or Fibroblasts: A Method to Characterize the Gastrointestinal Stem Cell NicheStem Cells Int., 2016 (2016), p. 3710836
View in ScopusGoogle Scholar - 230
H.N. Yan, A.S. Chan, F.P.-L. Lai, S.Y. Leung
Organoid cultures for cancer modelingCell Stem Cell, 30 (2023), pp. 917-937
View PDFView articleView in ScopusGoogle Scholar - 231
H. Lee, W. Hu, J.T. Matulay, M.V. Silva, T.B. Owczarek, K. Kim, C.W. Chua, L.J. Barlow, C. Kandoth, A.B. Williams, et al.
Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder CancerCell, 173 (2018), pp. 515-528.e17
View PDFView articleCrossRefGoogle Scholar - 232
Li, H.E. Francies, M. Secrier, J. Perner, A. Miremadi, N. Galeano-Dalmau, W.J. Barendt, L. Letchford, G.M. Leyden, E.K. Goffin, et al.
Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeuticsNat. Commun., 9 (2018), p. 2983
View at publisher
This article is free to access.CrossRefGoogle Scholar - 233
K. Dijkstra, C.M. Cattaneo, F. Weeber, M. Chalabi, J. van de Haar, L.F. Fanchi, M. Slagter, D.L. van der Velden, S. Kaing, S. Kelderman, et al.
Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor OrganoidsCell, 174 (2018), pp. 1586-1598.e12
View PDFView articleView in ScopusGoogle Scholar - 234
L. LeSavage, R.A. Suhar, N. Broguiere, M.P. Lutolf, S.C. Heilshorn
Next-generation cancer organoidsNat. Mater., 21 (2022), pp. 143-159
View at publisher CrossRefView in ScopusGoogle Scholar - 235
E. Ingber
Developmentally inspired human “organs on chips.”Development, 145 (2018), Article dev156125
View in ScopusGoogle Scholar - 236
Sontheimer-Phelps, B.A. Hassell, D.E. Ingber
Modelling cancer in microfluidic human organs-on-chipsNat. Rev. Cancer, 19 (2019), pp. 65-81
View at publisher CrossRefView in ScopusGoogle Scholar - 237
Ma, Y. Peng, H. Li, W. Chen
Organ-on-a-Chip: A New Paradigm for Drug DevelopmentTrends Pharmacol. Sci., 42 (2021), pp. 119-133
View PDFView articleView in ScopusGoogle Scholar - 238
R. Powley, M. Patel, G. Miles, H. Pringle, L. Howells, A. Thomas, C. Kettleborough, J. Bryans, T. Hammonds, M. MacFarlane, et al.
Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discoveryBr. J. Cancer, 122 (2020), pp. 735-744
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 239
Chiorazzi, J. Martinek, B. Krasnick, Y. Zheng, K.J. Robbins, R. Qu, G. Kaufmann, Z. Skidmore, M. Juric, L.A. Henze, et al.
Autologous humanized PDX modeling for immuno-oncology recapitulates features of the human tumor microenvironmentJ. Immunother. Cancer, 11 (2023), p. e006921
View at publisher CrossRefView in ScopusGoogle Scholar - 240
S. Thommen
Tumour avatars to model patients’ responses to immunotherapyNat. Rev. Cancer, 22 (2022), p. 660
View at publisher CrossRefView in ScopusGoogle Scholar - 241
M. Roelofsen, P. Voabil, M. de Bruijn, P. Herzig, A. Zippelius, T.N. Schumacher, D.S. Thommen
Protocol for ex vivo culture of patient-derived tumor fragmentsStar Protoc., 4 (2023), p. 102282
View PDFView articleView in ScopusGoogle Scholar - 242
Voabil, M. de Bruijn, L.M. Roelofsen, S.H. Hendriks, S. Brokamp, M. van den Braber, A. Broeks, J. Sanders, P. Herzig, A. Zippelius, et al.
An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancerNat. Med., 27 (2021), pp. 1250-1261
View at publisher CrossRefView in ScopusGoogle Scholar - 243
H. Dias, R. Bernards
Playing cancer at its own game: activating mitogenic signaling as a paradoxical interventionMol. Oncol., 15 (2021), pp. 1975-1985
View at publisher
This article is free to access.CrossRefView in ScopusGoogle Scholar - 244
H. Dias, A. Friskes, S. Wang, J.M. Fernandes Neto, F. van Gemert, S. Mourragui, H.J. Kuiken, S. Mainardi, D. Alvarez-Villanueva, C. Lieftink, et al.
Paradoxical activation of oncogenic signaling as a cancer treatment strategyPreprint at bioRxiv (2023), 10.1101/2023.02.06.527335
View at publisher Google Scholar - 245
M. van Neerven, N.E. de Groot, L.E. Nijman, B.P. Scicluna, M.S. van Driel, M.C. Lecca, D.O. Warmerdam, V. Kakkar, L.F. Moreno, F.A. Vieira Braga, et al.
Apc-mutant cells act as supercompetitors in intestinal tumour initiationNature, 594 (2021), pp. 436-441
View at publisher CrossRefView in ScopusGoogle Scholar - 246
Ge, E. Jakobsson
Systems Biology Understanding of the Effects of Lithium on CancerFront. Oncol., 9 (2019), p. 296
View in ScopusGoogle Scholar - 247
Bardia, J. Baselga
Neoadjuvant therapy as a platform for drug development and approval in breast cancerClin. Cancer Res., 19 (2013), pp. 6360-6370
View in ScopusGoogle Scholar - 248
U. Marron, M.D. Galsky, B. Taouli, M.I. Fiel, S. Ward, E. Kim, D. Yankelevitz, D. Doroshow, E. Guttman-Yassky, B. Ungar, et al.
Neoadjuvant clinical trials provide a window of opportunity for cancer drug discoveryNat. Med., 28 (2022), pp. 626-629
View at publisher CrossRefView in ScopusGoogle Scholar - 249
R. Reynolds, M. Moschetta, A.R. Yohannes, F. Walcott, M. Ashford, Z. Szucs, T. Sarbajna, J. Hadfield, E. Harrison, B.G. Challis, et al.
A View on Drug Development for Cancer PreventionCancer Discov., 13 (2023), pp. 1058-1083
View at publisher CrossRefView in ScopusGoogle Scholar - 250
T. Inan, P. Tenaerts, S.A. Prindiville, H.R. Reynolds, D.S. Dizon, K. Cooper-Arnold, M. Turakhia, M.J. Pletcher, K.L. Preston, H.M. Krumholz, et al.
Digitizing clinical trialsNPJ Digit. Med., 3 (2020), p. 101View in ScopusGoogle Scholar - 251
Alkhayyat, P. Kumar, K.O. Sanaka, P.N. Thota
Chemoprevention in Barrett’s esophagus and esophageal adenocarcinomaTherap. Adv. Gastroenterol., 14 (2021), Article 17562848211033730 - 252
Gale, K. Heider, A. Ruiz-Valdepenas, S. Hackinger, M. Perry, G. Marsico, V. Rundell, J. Wulff, G. Sharma, H. Knock, et al.
Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancerAnn. Oncol., 33 (2022), pp. 500-510
View PDFView articleView in ScopusGoogle Scholar - 253
Abbosh, A.M. Frankell, T. Harrison, J. Kisistok, A. Garnett, L. Johnson, S. Veeriah, M. Moreau, A. Chesh, T.L. Chaunzwa, et al.
Tracking early lung cancer metastatic dissemination in TRACERx using ctDNANature, 616 (2023), pp. 553-562
View at publisher CrossRefView in ScopusGoogle Scholar - 254
Powles, Z.J. Assaf, N. Davarpanah, R. Banchereau, B.E. Szabados, K.C. Yuen, P. Grivas, M. Hussain, S. Oudard, J.E. Gschwend, et al.
ctDNA guiding adjuvant immunotherapy in urothelial carcinomaNature, 595 (2021), pp. 432-437
View at publisher CrossRefView in ScopusGoogle Scholar - 255
Lipsyc-Sharf, E.C. de Bruin, K. Santos, R. McEwen, D. Stetson, A. Patel, G.J. Kirkner, M.E. Hughes, S.M. Tolaney, A.H. Partridge, et al.
Circulating Tumor DNA and Late Recurrence in High-Risk Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast cancerJ. Clin. Oncol., 40 (2022), pp. 2408-2419
View at publisher CrossRefGoogle Scholar - 256
Tie, J.D. Cohen, K. Lahouel, S.N. Lo, Y. Wang, S. Kosmider, R. Wong, J. Shapiro, M. Lee, S. Harris, et al.
Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer.N. Engl. J. Med., 386 (2022), pp. 2261-2272
View at publisher CrossRefView in ScopusGoogle Scholar - 257
André
Developing Anticancer Drugs in Orphan Molecular Entities – A Paradigm under ConstructionN. Engl. J. Med., 378 (2018), pp. 763-765
View at publisher CrossRefView in ScopusGoogle Scholar - 258
Andre, T. Filleron, M. Kamal, F. Mosele, M. Arnedos, F. Dalenc, M.P. Sablin, M. Campone, H. Bonnefoi, C. Lefeuvre-Plesse, et al.
Genomics to select treatment for patients with metastatic breast cancerNature, 610 (2022), pp. 343-348
View at publisher CrossRefView in ScopusGoogle Scholar - 259
Koh, D.J.H. Tan, C.H. Ng, C.E. Fu, W.H. Lim, R.W. Zeng, J.N. Yong, J.H. Koh, N. Syn, W. Meng, et al.
Patterns in Cancer Incidence Among People Younger Than 50 Years in the US, 2010 to 2019JAMA Netw. Open, 6 (2023), Article e2328171
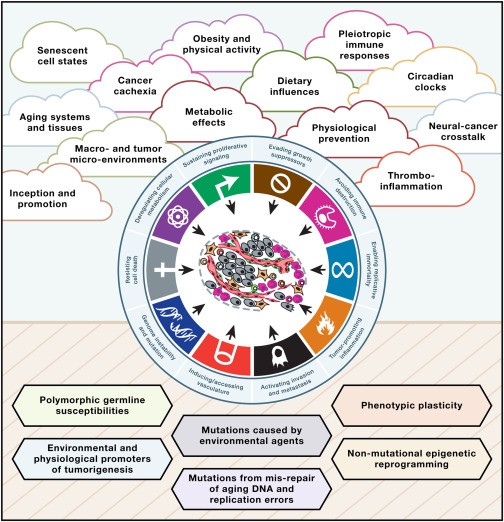
Figura 1. Las “nubes de complejidad” que inciden en las fronteras de la biología y la medicina del cáncer
Aunque las características distintivas del cáncer han proporcionado una justificación conceptual general para las innumerables manifestaciones que abarcan el cáncer como enfermedad, debajo de esta simplicidad se esconde una vertiginosa diversidad de efectos mecanicistas y fenotipos, tanto dentro de los tumores como a nivel de todo el sistema en el individuo afectado.
Por lo tanto, sobre el horizonte hay nubes de complejidad que, argumentamos desde esta perspectiva, son importantes y no se comprenden completamente. Debajo del horizonte se encuentran progresión del cáncer, que tampoco se comprenden completamente.
Esclarecer ambas dimensiones del cáncer como enfermedad sistémica será fundamental para lograr innovaciones revolucionarias en la prevención y el tratamiento duradero del cáncer humano.
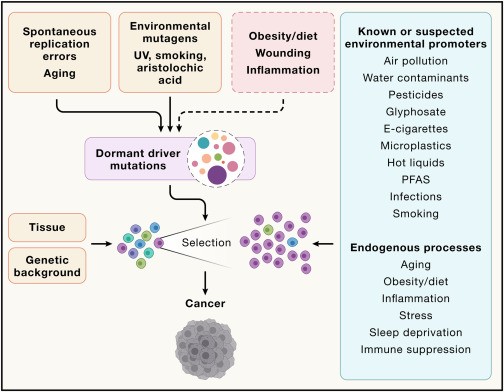
Figura 2. La interacción entre las mutaciones impulsoras del cáncer y los promotores tumorales ambientales o endógenos en el riesgo de cáncer
Las mutaciones son esenciales para el desarrollo de cánceres y pueden surgir como resultado de errores espontáneos en la replicación normal del ADN, durante el envejecimiento de células inactivas o por exposición a mutágenos en el medio ambiente.
La obesidad, los factores dietéticos y la inflamación también pueden contribuir indirectamente a la carga de mutaciones, por ejemplo, a través de la generación de especies reactivas de oxígeno (ROS), pero es poco probable que esto contribuya de manera importante a las cifras generales de mutaciones.
Las mutaciones pueden permanecer latentes en los tejidos normales durante largos períodos, a menos que el tejido se exponga repetidamente a un inductor de inflamación o lesión tisular, lo que provoca la selección de células portadoras de mutaciones específicas, lo que lleva a un crecimiento clonal.
Las mutaciones particulares seleccionadas dependen de muchos factores, incluido el tejido o célula de origen, los antecedentes genéticos del huésped o la naturaleza del factor promotor específico. Se muestran como ejemplos factores promotores exógenos conocidos o sospechados, así como posibles factores promotores endógenos o asociados al estilo de vida.
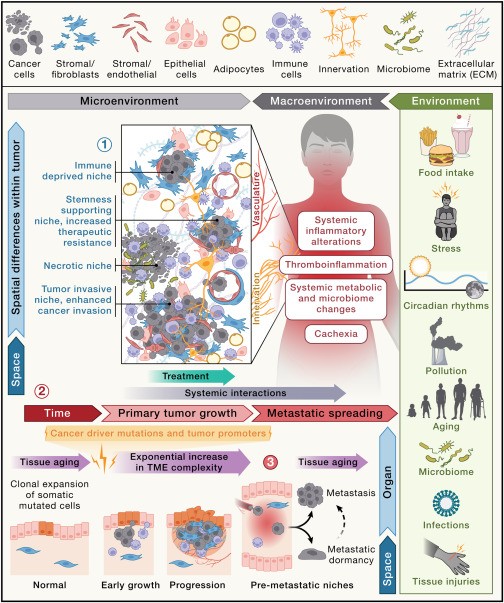
Figura 3. Microambientes y macroambientes tumorales.Representación del complejo ecosistema tumoral
Se representan las interacciones entre diversos componentes de los microambientes y macroambientes del tumor, que involucran influencias del huésped, así como factores externos, sobre el crecimiento del tumor y la diseminación metastásica.
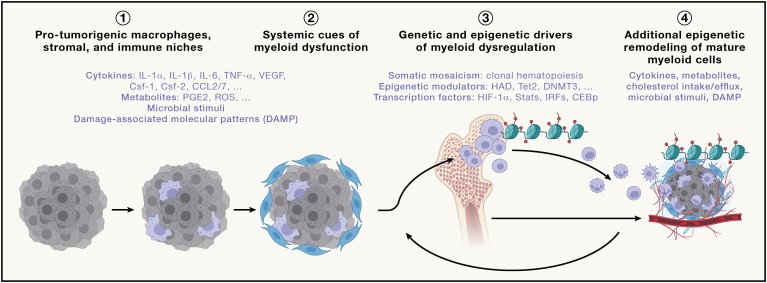
Figura 4. Impulsores locales y sistémicos de la desregulación mieloide protumorigénica
(1) Moléculas producidas durante el inicio del tumor que contribuyen al reclutamiento y activación de macrófagos que desempeñan un papel clave en el inicio de la cascada inflamatoria y la liberación de moléculas inflamatorias en la circulación sanguínea.
(2) Liberación de moléculas inflamatorias en la circulación sanguínea que impulsa la mejora y la desregulación de la mielopoyesis.
(3) Remodelación epigenética de progenitores mieloides en la médula ósea que contribuye a la inducción de nodos de programas supresores moleculares a lo largo del linaje mieloide.
(4) Impulsores adicionales producidos en el microambiente tumoral local que contribuyen a una mayor mejora de la supresión mieloide que amortigua aún más la inmunidad antitumoral y promueve la proliferación y el crecimiento de tumores. La lista de moléculas proporcionadas en cada categoría no es exhaustiva y solo se proporciona como ejemplos.
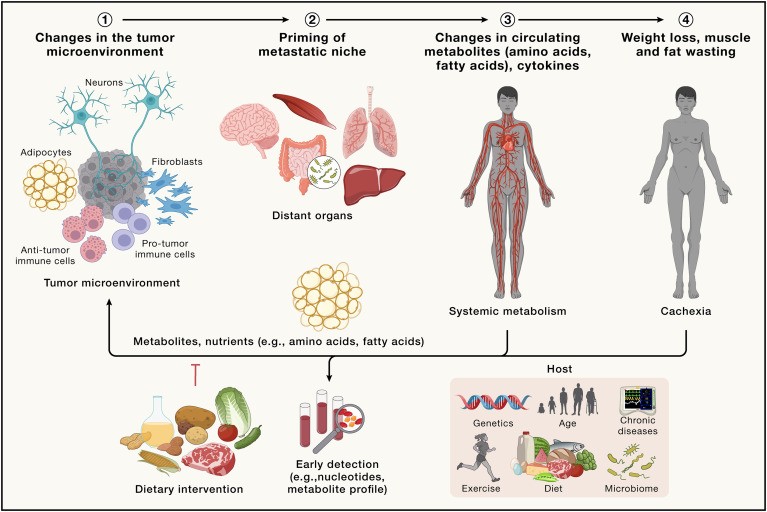
Figura 5. El cáncer como enfermedad sistémica
Relaciones recíprocas entre el metabolismo de las células cancerosas, el metabolismo de las células heterotípicas del microambiente tumoral y el metabolismo sistémico. Estas interferencias metabólicas modulan la progresión del cáncer y podrían conducir a caquexia asociada al cáncer. La detección de estas alteraciones metabólicas brinda oportunidades clínicas para la detección temprana y el seguimiento de la enfermedad.
Figura 6. Tromboinflamación y cáncer
La interacción entre los sistemas de coagulación y de inmunidad innata juega un papel importante durante el desarrollo y crecimiento de un tumor. Las interacciones tempranas entre los neutrófilos y las células tumorales, incluidas las trampas extracelulares de neutrófilos (NET) producidas por los neutrófilos, promueven el despertar de las células tumorales latentes, la supervivencia del tumor y el escape inmunológico.
Las agregaciones posteriores entre plaquetas y células tumorales promueven la supervivencia del tumor por vía intravascular y la diseminación del tumor. La activación de la coagulación a través de mecanismos específicos del tumor, como la liberación de factor tisular, aumenta el riesgo de trombosis asociada al cáncer.