
Ronald Palacios Castrillo, M.D.,PhD.
Resumen
Un número creciente de casos de pacientes apunta a un vínculo probable entre la infección por SARS-CoV-2 y la enfermedad de Parkinson (EP), pero aún se desconocen los mecanismos por los cuales el SARS-CoV-2 afecta al cerebro y genera síntomas neuropsiquiátricos en pacientes con COVID-19.
Ferroptosis, un tipo distinto de muerte celular no apoptótica dependiente del hierro caracterizado por peroxidación lipídica y agotamiento de glutatión, un factor clave en los trastornos neurológicos.
=> Recibir por Whatsapp las noticias destacadas
La ferroptosis puede tener un papel patogénico en la COVID-19, según hallazgos recientes; sin embargo, aún no se han investigado sus posibles contribuciones a la EP relacionada con la COVID-19. Aquí, analizamos las posibles vías de infección del cerebro por SARS-CoV-2.
Entre estos supuestos procesos, la ferroptosis puede contribuir a la etiología de la EP asociada a la COVID-19, proporcionando potencialmente métodos terapéuticos.
En Detalle
La epidemia del síndrome respiratorio agudo severo por coronavirus 2 (SARS-CoV-2), conocida como la pandemia de la nueva enfermedad por coronavirus de 2019 (COVID-19), ha provocado ansiedad global y una catástrofe económica [1].
Hasta el 17 de diciembre de 2023, se han notificado a la Organización Mundial de la Salud 772 millones de casos confirmados de COVID-19 en todo el mundo, incluidas 6,9 millones de muertes [2].
El impacto de la COVID-19 ha sido insuperable hasta ahora y sus efectos a largo plazo podrían ser mucho más desastrosos [3, 4]. El virus SARS-CoV-2, causante de la actual pandemia de COVID-19, afecta no sólo al sistema respiratorio, sino que también afecta a otros órganos y tejidos [5]. El SARS-CoV-2 se ha descubierto recientemente en neuronas de varias partes del cerebro, incluida la sustancia negra [6, 7].
Varias personas con infecciones por SARS-CoV-2 han informado haber experimentado complicaciones neurológicas agudas y subagudas [8,9,10]. Debido a una serie de causas que provocan una reducción de las neuronas dopaminérgicas de la sustancia negra seguida de un agotamiento de la dopamina estriatal, los pacientes con enfermedad de Parkinson (EP) tienen una variedad de alteraciones motoras y no motoras [11]. En esta revisión, exploramos la evidencia actual que indica posibles vínculos patogénicos entre COVID-19 y la EP, y brindamos direcciones para posibles enfoques terapéuticos con efásis en la ferroptosis.
COVID-19 y EP: un panorama más definido
Si bien aún se desconoce el mecanismo específico que causa la presunta degradación de las neuronas dopaminérgicas nigroestriatales después de una infección viral, la infección viral está recibiendo cada vez más atención como causa de la EP [12, 13].
Los estudios han indicado que el SARS-CoV-2 puede infiltrarse en el sistema nervioso central (SNC) y causar disfunción neurológica adicional en un porcentaje considerable de pacientes infectados [9, 14].
El 36% de las infecciones por SARS-CoV-2 experimentan síntomas neurológicos durante la etapa aguda, el 25% de los cuales pueden estar relacionados directamente con la afectación del SNC [15]. La sustancia negra es particularmente susceptible al SARS-CoV-2 [16].
No todas las poblaciones neuronales son igualmente propensas a la degeneración. Debido a sus características intrínsecas, como las altas necesidades de energía para soportar una mayor fosforilación oxidativa basal en las mitocondrias, una alta densidad terminal axónica y una arborización axonal sustancial, las neuronas dopaminérgicas son particularmente susceptibles a la degeneración.
Curiosamente, al menos 20 casos muestran que los pacientes con COVID-19 experimentaron parkinsonismo clínico después de la infección por SARS-CoV-2 [17], lo que apunta a un vínculo potencial entre la infección por COVID-19 y el parkinsonismo recién formado.
Neurotropismo de COVID-19 y EP: explorando los vínculos
El SNC se ha visto alterado por el COVID-19 de diversas formas, incluida la invasión directa del SARS-CoV-2 a las células neuronales, enormes elementos inflamatorios impulsados por una inflamación sistémica grave que fluye hacia el cerebro, insuficiencia respiratoria relacionada con la isquemia cerebral, etc. [18,19 ,20].
Bulbo olfatorio
Hasta el 20% de los adultos con personas infectadas por COVID-19 presentan anosmia/hiposmia y ageusia, que es un síntoma neurológico, en una etapa temprana de la enfermedad viral [21]. La anosmia, sin embargo, es una señal precursora bien conocida del desarrollo de la EP [22].
Además, los estudios demostraron que el SARS-CoV-2 puede ingresar directamente a través de las neuronas olfativas y, curiosamente, sin afectar primero los pulmones [23].
La neurogénesis deteriorada en el sistema olfativo puede afectar la anosmia en COVID-19 y la EP [24]. El SARS-CoV-2 puede tener acceso directo a áreas del cerebro para el desarrollo de la EP, según investigaciones neuropatológicas que utilizan inmunotinción de agregados de α-sinucleína que implican que la EP comienza en las neuronas olfatorias o intestinales y progresa hasta el cerebro.
Aparentemente, el SARS-CoV-2 puede ingresar al cerebro a través de las vías olfativas y extenderse a los ganglios basales, el tronco encefálico y la corteza piriforme e infralímbica [25].
Microbioma intestinal y fisiología intestinal.
Un estado inflamatorio sistémico que induce el SARS-CoV-2 puede aumentar el riesgo de EP además de la invasión directa del SNC [26].
El COVID-19 también provoca síntomas gastrointestinales, y se ha encontrado RNA del SARS-CoV-2 en las heces de personas infectadas, lo que sugiere que el virus es de origen intestinal.
Según un estudio reciente [25], los enterocitos son las principales células diana del SARS-CoV-2 y responden a la infección desencadenando una potente respuesta inflamatoria. Estos resultados podrían enfatizar aún más la posible función de COVID-19 como factor de riesgo de EP [27].
Otra idea sostiene que la microbiota intestinal es el punto de partida del proceso inflamatorio que resulta en la EP [28].
Sorprendentemente, los síntomas neurológicos y los cambios en la microbiota intestinal observados en pacientes con COVID-19 también están presentes con frecuencia en pacientes con EP [29].
Además, la infección intestinal por SARS-CoV-2 puede cambiar la fisiología intestinal en general y la microbiota intestinal [30], impactando todos los aspectos que “periféricamente” contribuyen a la etiología y el desarrollo de la EP [31].
Enzima convertidora de angiotensina 2 (ACE2)
Uno de los principales receptores que facilita la entrada del SARS-CoV-2 en las células humanas es el receptor ACE2 [32, 33]. Después de la infección, COVID-19 tiene una mayor afinidad por la proteína S, lo que permite que la glicoproteína viral se una a las células huésped ACE2 [34,35,36].
Estos receptores están ampliamente distribuidos en las neuronas y las células gliales de muchas áreas del cerebro, incluida la corteza cerebral, el cuerpo estriado, la sustancia negra y el tronco del encéfalo [37].
En las neuronas dopaminérgicas, que están disminuidas en los pacientes con EP, la ACE2 se expresa significativamente y puede contribuir al agravamiento de síntomas preexistentes o una infección por COVID-19 más grave [38].
Debido a que ACE2 y DOPA descarboxilasa coexpresan y coregulan en tipos de células no neuronales, la ruta de síntesis de dopamina puede estar implicada en la patogénesis de COVID-19 [39, 40].
La expresión de ACE2 está regulada a la baja por la infección por SARS-CoV, lo que puede contribuir al deterioro de la producción de dopamina [41,42,43].
Existe evidencia de que los niveles de expresión de ACE2 en el cerebro de pacientes con EP se han reducido, provocando pérdida y degeneración de neuronas dopaminérgicas [44,45,46].
COVID-19 y EP: vías inflamatorias compartidas bajo estrés oxidativo
El SARS-CoV-2 tiene la capacidad de generar una desregulación de las citocinas: una “tormenta de citocinas” [47]. Para regular la infección que podría dañar las neuronas, las neuronas infectadas liberan citocinas como el receptor de interleucina-2, la interleucina-6 y el factor de necrosis tumoral [48].
El desarrollo tanto de COVID-19 como de EP puede verse influenciado significativamente por el estrés oxidativo y la tormenta de citocinas.
Además, la barrera hematoencefálica (BHE) puede romperse como resultado de la grave respuesta inflamatoria sistémica provocada por una infección viral. Como resultado, las citoquinas periféricas pueden ingresar al SNC, donde pueden causar o exacerbar la neuroinflamación [49].
Se cree que la inflamación inducida por virus contribuye a la neurodegeneración [50], al igual que el daño por «múltiples golpes» [51]. Al igual que el concepto de “dos golpes” de la EP, la infección por COVID-19 podría haber servido como un segundo golpe infeccioso [52].
La respuesta inflamatoria inducida por una infección aguda o crónica puede iniciar o acelerar procesos tempranos y subclínicos subyacentes a las primeras etapas de la EP.
Además, la investigación sobre enfermedades neurodegenerativas y otras infecciones virales indica que la inflamación sistémica provocada por la infección por SARS-CoV-2 puede contribuir aún más a los procesos neuroinflamatorios y aumentar la susceptibilidad a la EP [53].
Además, el descubrimiento de posibles estrategias terapéuticas para el tratamiento de la COVID-19 y la EP se ve favorecido por la supresión dirigida de las caspasas y la activación del factor nuclear kappa B [54].
Debido a las propiedades antiinflamatorias de la vitamina D3, la suplementación regular con 2000-5000 UI/día de D3 puede ayudar a las personas mayores con EP a reducir la evolución de su enfermedad y también puede proporcionar una mayor protección contra la COVID-19 [55].
α-sinucleína
El sistema dopaminérgico nigroestriatal sufre de neurodegeneración provocada por la α-sinucleína, que es clínicamente evidente como los síntomas habituales de la EP/parkinsonianos.
Se cree que la sobreexpresión de α-sinucleína está relacionada con la infección por SARS-CoV-2. La neuroinfección por SARS-CoV-2 provoca niveles elevados de α-sinucleína [46, 47]. De hecho, la infección por SARS-CoV-2 parece provocar la agregación de α-sinucleína en el cerebro de los casos de COVID-19 [56]. Experimentos in vitro han demostrado que la proteína N del SARS-CoV-2 acelera la agregación de α-sinucleína [57].
La microinyección de proteína N interrumpió la proteostasis de la α-sinucleína y aumentó la mortalidad celular en las células SH-SY5Y [57].
Además, la infección por SARS-CoV-2 puede dificultar potencialmente la eliminación de la α-sinucleína. La sobreexpresión de α-sinucleína, que puede desempeñar un papel en la respuesta inmune [58], puede provocar que la microglía se active [59].
Las células de microglía amplificarían la respuesta inflamatoria y liberarían citocinas y quimiocinas inflamatorias, lo que provocaría la muerte neuronal [60, 61].
Además, la excitotoxicidad del glutamato, que está relacionada con la degeneración neuronal, puede ser el resultado de reacciones neuroinmunes a una infección [62, 63]. Por lo tanto, la infección por SARS-CoV-2 parece afectar la α-sinucleína y la muerte de las neuronas dopaminérgicas, que se sabe que causa la EP.
Células gliales
Actualmente se considera que los astrocitos y la microglía, en particular, desempeñan un papel importante en las respuestas tanto beneficiosas como negativas del huésped durante los estados de enfermedad del SNC [64].
Con el creciente número de personas infectadas y reinfectadas en todo el mundo, la microglía puede desempeñar un papel en la fisiopatología de las enfermedades neurológicas posteriores a la COVID-19, incluida la EP [65].
Mediante la regulación positiva de los genes de citocinas inflamatorias y la mayor permeabilidad de la BHE, los astrocitos reactivos participan con frecuencia en procesos de neurodegeneración y neuroinflamación.
Cuando las células lesionadas detectan ligandos endógenos o derivados de patógenos, los receptores de reconocimiento de patrones (PRR), que son producidos por los astrocitos y la microglía, inician la respuesta inmune innata [66].
Los receptores tipo peaje (TLR), una clase bien conocida de PRR, podrían estar involucrados en la tormenta de citoquinas provocada por el SARS-CoV-2 [67].
De hecho, es probable que los TLR4 detecten patrones moleculares derivados del SARS-CoV-2 y desencadenen una respuesta inflamatoria. TLR2 y TLR7/TLR8 también son activados por el SARS-CoV-2 [68].
Puede surgir una tormenta de citocinas en el SNC a partir de la activación simultánea de varios TLR. Los TLR contribuyen a la EP mediando la neuroinflamación y la activación glial asociadas [69].
La interacción entre la α-sinucleína y el TLR2 microglial promueve el crecimiento y la propagación de la patología de la α-sinucleína [69].
Estrés del retículo endoplásmico y mitocondrias.
Como el SARS-CoV-2 inhibió simultáneamente la expresión de las selenoproteinas SELENOF, SELENOM, SELENOK y SELENOS, el retículo endoplasmático es un orgánulo gravemente dañado por el virus.
El estrés del retículo endoplásmico y la respuesta de la proteína desplegada están impulsados por la replicación del coronavirus en las células infectadas [70,71,72,73].
Aunque se sabe que las selenoproteínas residentes en el retículo endoplásmico desempeñan un papel en la preservación de la homeostasis del retículo endoplásmico, aún no se ha establecido una conexión entre la infección por coronavirus y las selenoproteínas residentes en el retículo endoplásmico [74].
Además, las mitocondrias desempeñan un papel en la inducción de la respuesta inflamatoria, incluida la producción de especies reactivas de oxígeno (ROS) mitocondriales y la regulación positiva de la expresión de genes vinculados a enzimas relacionadas con la glucólisis, algo que también se ha informado ampliamente en el SNC en COVID-19 [75].
La desregulación del eje mitocondrial ACE2/MrgE/NO puede tener un efecto significativo en los procesos neurodegenerativos de las neuronas dopaminérgicas, donde la disfunción mitocondrial y el estrés oxidativo pueden tener un impacto sustancial [76].
Firma de ferroptosis en la infección por SARS-CoV-2 y mecanismos moleculares de la ferroptosis
Un estudio de caso de un paciente con COVID-19 mostró la presencia de una firma de ferroptosis en los tejidos cardíaco y renal [77]. El hallazgo fue el primero en documentar una firma de ferroptosis en COVID-19, que se pensaba que era un factor de riesgo de daño orgánico.
Además, una investigación in vitro reveló que el SARS-CoV-2 redujo la glutatión peroxidasa 4 (GPX4), que era el freno de la ferroptosis [78]. El SARS-CoV-2 infecta fácilmente las células marcapasos, lo que da como resultado una tasa notablemente aumentada de ferroptosis [79].
Un creciente conjunto de investigaciones ha revelado que la ferroptosis desempeña importantes funciones patogénicas en el cáncer, el daño orgánico por isquemia y la demencia desde que fue utilizada por primera vez por Dixon et al. [80].
El mecanismo preciso que subyace a la ferroptosis aún se desconoce, pero se sabe que la alteración del metabolismo del hierro, el agotamiento del glutatión (GSH), la inactivación de GPX4 y el aumento de la peroxidación de PUFA por ROS desempeñan papeles clave en su inicio y progresión (80, 81).
En general, la sobrecarga de hierro en las células, la disminución de la expresión de GPX4 y xCT, la activación del miembro 4 de la familia de cadena larga de la acilCoA sintetasa (ACSL4) y la lisofosfatidilcolina aciltransferasa-3, y un aumento en la peroxidación lipídica son los cuatro mecanismos principales que inducen la ferroptosis [82 ,83,84].
El papel potencial de la ferroptosis subyace a la EP relacionada con COVID-19. La ferroptosis puede existir en la EP relacionada con COVID-19
Un aspecto potencialmente letal de la infección por SARS-CoV-2 es la implicación de síntomas neuropsiquiátricos, como se mencionó anteriormente.
La ferroptosis se ha identificado como un mecanismo clave para la muerte de las neuronas dopaminérgicas en la EP [85].
El deterioro grave del comportamiento y la muerte neuronal de ratones que recibieron el inhibidor de la ferroptosis ferrostatina-1 24 horas antes de 1-metil-4-fenil-1, 2, 3, 6-tetrahidropiridina (MPTP) se revirtieron en gran medida [86].
La ferrostatina-1 también tiene un impacto neuroprotector en las células SH-SY5Y dañadas por rotenona y 1-metil-4-fenilpiridinio (MPP+) [87, 88], lo que sugiere que la ferroptosis podría ofrecer una alternativa para el tratamiento de la EP.
Los análogos mejorados de ferrostatina-1 y liproxstatina-166, dos miembros de la nueva generación de inhibidores de la ferroptosis, pueden usarse para evaluar la participación de la ferroptosis en la infección por SARS-CoV-2, así como para tratar potencialmente la COVID-19 [89, 90 ].
Desregulación del metabolismo del hierro en la EP relacionada con COVID-19
La alteración del metabolismo del hierro, un importante contribuyente a la EP [91, 92], se ha establecido ampliamente en una fracción significativa de pacientes con COVID-19 en respuesta a la infección por SARS-CoV-2 [93,94,95], lo que se corresponde con el riesgo de enfermedad grave y mortal por COVID-19.
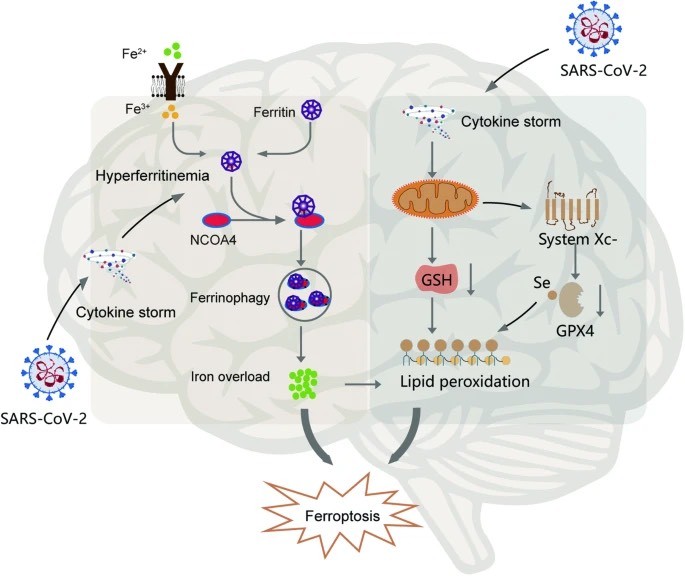
En nuestro estudio anterior, elaboramos el mecanismo de desregulación del metabolismo del hierro y la ferritinofagia en COVID-19 [96, 97].
Además, los niveles de ceruloplasmina en pacientes con COVID-19 a largo plazo muestran una tendencia a la baja en comparación con los de pacientes con COVID-19 y controles sanos [98].
La EP es causada en parte por la neurotoxicidad de la acumulación de hierro provocada por una actividad ferroxidasa inadecuada o reducida de la ceruloplasmina [99, 100].
La acumulación de hierro puede causar un aumento en la reserva intracelular de hierro lábil (II) y la reacción de Fenton, lo que resulta en la producción de ROS lipídicos y ferroptosis.
El agotamiento del hierro intracelular sería una posible opción de tratamiento para la COVID-19. Se ha demostrado que la deferoxamina y el imatinib previenen la infección de las células marcapasos por SARS-CoV-2, así como la ferroptosis inducida por la infección por SARS-CoV-2 [79].
Eje GSH-GPX4 en la EP relacionada con COVID-19
La producción mitocondrial de ROS aumentó por la infección por SARS-CoV-2 y su replicación [101]. GPX4, ubicado en las mitocondrias, protege específicamente contra la muerte celular ferroptótica.
La expresión del gen GPX4 es suprimida por el SARS-CoV-2, lo que promovió la aparición de ferroptosis. Una investigación fundamental que infectó células de riñón de mono verde africano (Vero) con SARS-CoV2 derivado de pacientes descubrió que los niveles de mRNA de GPX4 estaban considerablemente reducidos, lo que sugiere una conexión entre la ferroptosis y el SARS-CoV-2 [78].
La leucopenia en pacientes con COVID-19 puede estar relacionada con la ferroptosis en los leucocitos y la supresión de GPX4 causada por el SARS-CoV-2 [102].
La falta de GPX4 indujo la pérdida de la capacidad de peroxidación del GSH para minimizar las ROS lipídicas producidas por la reacción de Fenton. Por lo tanto, la peroxidación lipídica y la ferroptosis se producirían por una acumulación de ROS lipídicos. En consecuencia, es probable que la ferroptosis contribuya a los síntomas de EP de COVID-19
(Fig. 1).
Legenda Figura 1. La infección por SARS-CoV-2 puede provocar ferroptosis en la patogénesis de la EP, lo que probablemente contribuya al inicio de la enfermedad a través de dos vías principales: la vía dependiente del transportador con desequilibrio de hierro y la vía intrínseca o regulada por enzimas.
La infección por SARS-CoV-2 puede provocar ferroptosis en la patogénesis de la EP, lo que probablemente contribuya al inicio de la enfermedad a través de dos vías principales: la vía dependiente del transportador con desequilibrio de hierro y la vía intrínseca o regulada por enzimas.
Desafíos para establecer relaciones causales entre COVID-19 y EP
Aún es necesario abordar muchas preocupaciones, a pesar de que están comenzando a surgir numerosas investigaciones relacionadas sobre la COVID-19 y la EP.
No está claro si distintas cepas de SARS-CoV-2 causan diferentes síntomas neurológicos. Para determinar la conexión entre las mutaciones del SARS-CoV-2 y las manifestaciones de la EP, se requieren análisis más profundos. Además, se ha descubierto que varias formas de muerte celular, como la autofagia, la apoptosis y la piroptosis, están implicadas en el mecanismo patogénico tanto de la COVID-19 como de la EP [103, 104].
Dado que actualmente es bastante difícil determinar la interacción entre las diferentes vías de muerte celular en la EP relacionada con COVID-19, en esta revisión no se explora ninguno de los mecanismos de muerte celular mencionados anteriormente, aparte de la ferroptosis.
Además, no se evalúan la edad y el sexo de la víctima de la infección por SARS-CoV-2, que podrían ser variables que contribuyen al desarrollo de la EP. Además, sería necesario tener en cuenta las influencias tanto ambientales como genéticas.
Tanto la invasión neuronal directa como los efectos indirectos de la neuroinflamación pueden estar involucrados en los síntomas neuropsiquiátricos de la COVID-19. Será posible lograr una comprensión más profunda y una caracterización funcional de la EP relacionada con COVID-19 mediante el uso de evaluaciones de alto rendimiento y organoides derivados de pacientes, que pueden proporcionar un medio viable para dilucidar indicios fisiopatológicos y posibles enfoques de tratamiento.
Las posibles implicaciones clínicas de la ferroptosis en la EP relacionada con COVID-19
Como nueva forma de muerte celular, la ferroptosis es muy prometedora para el estudio de la COVID-19 asociada a la EP.
Un posible enfoque de tratamiento podría implicar centrarse en la ferroptosis. Hasta ahora, los quelantes de hierro y los antioxidantes lipófilos han sido los principales enfoques para suprimir la ferroptosis [105]. Mediante el control de la reacción de Fenton, los quelantes del hierro como la deferoxamina quelan el hierro y detienen la peroxidación lipídica.
Es importante señalar que las alteraciones del hierro en el cerebro pueden comprobarse fácilmente mediante un mapeo de susceptibilidad cuantitativa [106]. La ferrostatina-1 y la lipoxstatina-1 son antioxidantes lipófilos típicos que eliminan los peróxidos lipídicos e inhiben la ferroptosis.
Los estudios han demostrado que tanto los quelantes de hierro como los antioxidantes lipófilos podrían prevenir la progresión de la EP [107]. Además, la deferoxamina reduce las cantidades de IL-6, una importante citoquina inflamatoria generada durante la COVID-19 [108], lo que sugiere que la deferoxamina puede usarse como medicamento para tratar la EP inducida por la COVID-19.
Sin embargo, ningún estudio ha evaluado aún la eficacia de los antioxidantes lipófilos en el proceso de terapia COVID-19. Además, las investigaciones futuras deberían analizar los posibles beneficios de combinar citocinas antiinflamatorias con interferencia de ferroptosis para mejorar la resiliencia de los pacientes con EP relacionados con la COVID-19, teniendo en cuenta que se cree que las tormentas de citocinas inflamatorias son contribuyentes clave a la COVID-19.
Hasta donde sabemos, no existen ensayos clínicos que evalúen los inhibidores de la ferroptosis en la EP relacionada con COVID-19 ni ninguna indicación de la firma de ferroptosis en los tejidos cerebrales de los pacientes con COVID-19.
Aunque hemos logrado grandes avances en la comprensión del papel patogénico de la ferroptosis en la EP, no está claro el papel preciso que desempeña la ferroptosis en el cerebro dañado por el SARS-CoV-2 y cómo inicia la inflamación que finalmente causa daño cerebral.
Además, es difícil describir que la ferroptosis es un efecto secundario de la infección por SARS-CoV-2 o si es una forma en que el virus se replica y se vuelve más peligroso durante el COVID-19.
A pesar de que hemos dilucidado la conexión entre tres vías primarias de ferroptosis y COVID-19, ¿qué vía es más importante en el daño cerebral relacionado con COVID-19? Es necesario abordar estas cuestiones para poder presentar un argumento completo y convincente a favor del uso terapéutico de los inhibidores de la ferroptosis.
Conclusión
Desde el comienzo de la epidemia, los científicos han estado trabajando febrilmente para descubrir una nueva vacuna contra el COVID-19 o un posible tratamiento. Esta revisión generalmente analiza la relación entre COVID-19 y la EP (Fig. 2).

Legenda Figura 2.
El SARS-CoV-2 podría infiltrarse en el SNC directamente a través del olfato, el tracto respiratorio, el tracto gastrointestinal y la barrera hematoencefálica. La infección podría provocar agregación citotóxica de α-sinucleína, estrés del retículo endoplásmico, daño a las mitocondrias, neuroinflamación y ferroptosis, que inducen la degeneración de las neuronas dopaminérgica.
El curso de la COVID-19 y la EP muestra similitudes en algunos procesos bioquímicos, incluido el estrés oxidativo, la inflamación y la agregación de proteínas [53].
Dado que el COVID-19 presenta síntomas inusuales que incluyen agotamiento de GSH, inactivación de GPX4, metabolismo anormal del hierro y elevación de la peroxidación de PUFA por especies reactivas de oxígeno, es posible que el SARS-CoV-2 pueda causar ferroptosis en las neuronas dopaminérgicas, lo que luego contribuiría al desarrollo de EP.
Por lo tanto, esta revisión presenta evidencia de que la ferroptosis está íntimamente relacionada y es muy prometedora para la investigación sobre la EP relacionada con COVID-19, lo que proporciona una dirección de investigación prometedora.
Especulamos que la ferroptosis contribuye a la EP relacionada con la infección por SARS-CoV-2 a la luz del posible vínculo entre la ferroptosis y las anomalías neurológicas en pacientes con COVID-19.
Aunque falta evidencia sobre estrategias de tratamiento efectivas para la EP relacionada con COVID-19, una táctica potencialmente efectiva sería atacar la ferroptosis.
Sin embargo, aún se desconoce cómo funciona la ferroptosis en las neuronas dopaminérgicas infectadas con SARS-CoV-2 o si representa un nuevo objetivo terapéutico prometedor para la terapia de EP relacionada con COVID-19.
Se necesitan urgentemente más investigaciones para confirmar que la ferroptosis ocurre en el COVID-19, aclarar su mecanismo preciso y determinar si está relacionada con el daño cerebral asociado con el COVID-19.
Referencias Bibliográficas
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–74.
Article
CAS
PubMed
PubMed Central
Google Scholar
2. World Health Organization. 2023. https://covid19.who.int.
Guo D, Han B, Lu Y, Lv C, Fang X, Zhang Z, et al. Influence of the COVID-19 pandemic on quality of life of patients with Parkinson’s disease. Parkinsons Dis. 2020;2020:1216568.
PubMed
PubMed Central
Google Scholar
3. Koelle K, Martin MA, Antia R, Lopman B, Dean NE. The changing epidemiology of SARS-CoV-2. Science. 2022;375:1116–21.
Article
ADS
CAS
PubMed
PubMed Central
Google Scholar
4. Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023;21:133–46.
Article
CAS
PubMed
PubMed Central
Google Scholar
5. Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med. 2021;218:e20202135.
Article
CAS
PubMed
PubMed Central
Google Scholar
6. Emmi A, Rizzo S, Barzon L, Sandre M, Carturan E, Sinigaglia A, et al. Detection of SARS-CoV-2 viral proteins and genomic sequences in human brainstem nuclei. NPJ Parkinsons Dis. 2023;9:25.
Article
CAS
PubMed
PubMed Central
Google Scholar
7. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. 2020;143:3104–20.
Article
PubMed
PubMed Central
Google Scholar
8. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77:683–90.
Article
PubMed
Google Scholar
9. Taquet M, Geddes JR, Husain M, Luciano S, Harrison PJ. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry. 2021;8:416–27.
Article
PubMed
PubMed Central
Google Scholar
10. Grayson M. Parkinson’s disease. Nature. 2016;538:S1.
Article
ADS
CAS
PubMed
Google Scholar
11. Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. 2020;36:1–12.
Article
PubMed
Google Scholar
12. Pang SY, Ho PW, Liu HF, Leung CT, Li L, Chang EES, et al. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodegener. 2019;8:23.
Article
PubMed
PubMed Central
Google Scholar
13. Asadi-Pooya AA, Simani L. Central nervous system manifestations of COVID-19: a systematic review. J Neurol Sci. 2020;413:116832.
Article
CAS
PubMed
PubMed Central
Google Scholar
14. Heneka MT, Golenbock D, Latz E, Morgan D, Brown R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res Ther. 2020;12:69.
Article
CAS
PubMed
PubMed Central
Google Scholar
15. Merello M, Bhatia KP, Obeso JA. SARS-CoV-2 and the risk of Parkinson’s disease: facts and fantasy. Lancet Neurol. 2021;20:94–95.
Article
CAS
PubMed
Google Scholar
16. Boura I, Chaudhuri KR. Coronavirus disease 2019 and related Parkinsonism: the clinical evidence thus far. Mov Disord Clin Pract. 2022;9:584–93.
Article
PubMed
PubMed Central
Google Scholar
17. Soung AL, Vanderheiden A, Nordvig AS, Sissoko CA, Canoll P, Mariani MB, et al. COVID-19 induces CNS cytokine expression and loss of hippocampal neurogenesis. Brain. 2022;145:4193–201.
Article
PubMed
PubMed Central
Google Scholar
18. Monje M, Iwasaki A. The neurobiology of long COVID. Neuron. 2022;110:3484–96.
Article
CAS
PubMed
PubMed Central
Google Scholar
19. Krasemann S, Haferkamp U, Pfefferle S, Woo MS, Heinrich F, Schweizer M, et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. 2022;17:307–20.
Article
CAS
Google Scholar
20. Cocco A, Amami P, Desai A, Voza A, Ferreli F, Albanese A. Neurological features in SARS-CoV-2-infected patients with smell and taste disorder. J Neurol. 2021;268:1570–72.
Article
CAS
PubMed
Google Scholar
21. Schaeffer E, Postuma RB, Berg D. Prodromal PD: a new nosological entity. Prog Brain Res. 2020;252:331–56.
Article
PubMed
Google Scholar
22. Li K, Wohlford-Lenane C, Perlman S, Zhao J, Jewell AK, Reznikov LR, et al. Middle east respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J Infect Dis. 2016;213:712–22.
Article
CAS
PubMed
Google Scholar
23. Rethinavel HS, Ravichandran S, Radhakrishnan RK, Kandasamy M. COVID-19 and Parkinson’s disease: Defects in neurogenesis as the potential cause of olfactory system impairments and anosmia. J Chem Neuroanat. 2021;115:101965.
Article
CAS
PubMed
PubMed Central
Google Scholar
24. Lechien JR, Chiesa-Estomba CM, De Siati DR, Horoi M, Le Bon SD, Rodriguez A, et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Otorhinolaryngol. 2020;277:2251–61.
Article
PubMed
PubMed Central
Google Scholar
25. Krashia P, Cordella A, Nobili A, La Barbera L, Federici M, Leuti A, et al. Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat Commun. 2019;10:3945.
Article
ADS
PubMed
PubMed Central
Google Scholar
26. Zhou J, Li C, Liu X, Chiu MC, Zhao X, Wang D, et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. 2020;26:1077–83.
Article
CAS
PubMed
Google Scholar
27. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469–1480.e12.
Article
CAS
PubMed
PubMed Central
Google Scholar
28. Sun MF, Shen YQ. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s Disease. Ageing Res Rev. 2018;45:53–61.
Article
CAS
PubMed
Google Scholar
29. Dhar D, Mohanty A. Gut microbiota and Covid-19- possible link and implications. Virus Res. 2020;285:198018.
Article
CAS
PubMed
Google Scholar
30. Borghammer P. How does parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov Disord. 2018;33:48–57.
Article
PubMed
Google Scholar
31. Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11:995–98.
Article
CAS
PubMed
Google Scholar
32. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–4.
Article
ADS
CAS
PubMed
PubMed Central
Google Scholar
33. Papa SM, Brundin P, Fung VSC, Kang UJ, Burn DJ, Colosimo C, et al. Impact of the COVID-19 pandemic on Parkinson’s disease and movement disorders. Mov Disord. 2020;35:711–15.
Article
CAS
PubMed
PubMed Central
Google Scholar
34. Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. 2020;92:552–55.
Article
CAS
PubMed
PubMed Central
Google Scholar
35. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–63.
Article
ADS
CAS
PubMed
PubMed Central
Google Scholar
36. Chen X, Laurent S, Onur OA, Kleineberg NN, Fink GR, Schweitzer F, et al. A systematic review of neurological symptoms and complications of COVID-19. J. Neurol. 2021;268:392–402.
Article
CAS
PubMed
Google Scholar
37. Brown EG, Chahine LM, Goldman SM, Korell M, Mann E, Kinel DR, et al. The effect of the COVID-19 pandemic on people with Parkinson’s disease. J. Parkinsons Dis. 2020;10:1365–77.
Article
CAS
PubMed
PubMed Central
Google Scholar
38. Nataf S. An alteration of the dopamine synthetic pathway is possibly involved in the pathophysiology of COVID-19. J. Med Virol. 2020;92:1743–44.
Article
CAS
PubMed
PubMed Central
Google Scholar
39. Orru G, Conversano C, Malloggi E, Francesconi F, Ciacchini R, Gemignani A. Neurological complications of COVID-19 and possible neuroinvasion pathways: a systematic review. Int J Environ Res Public Health. 2020;17:6688.
Article
CAS
PubMed
PubMed Central
Google Scholar
40. Khalefah MM, Khalifah AM. Determining the relationship between SARS-CoV-2 infection, dopamine, and COVID-19 complications. J Taibah Univ Med Sci. 2020;15:550–53.
PubMed
PubMed Central
Google Scholar
41. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9.
Article
CAS
PubMed
PubMed Central
Google Scholar
42. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20.
Article
CAS
PubMed
PubMed Central
Google Scholar
43. Achbani A, Sine H, Naciri A, Baba MA, Kharbach A, Bouchriti Y, et al. Can the 2019 novel coronavirus cause Parkinson’s disease? Mov Disord. 2020;35:1102–03.
Article
CAS
PubMed
PubMed Central
Google Scholar
44. Pavel A, Murray DK, Stoessl AJ. COVID-19 and selective vulnerability to Parkinson’s disease. Lancet Neurol. 2020;19:719.
Article
CAS
PubMed
PubMed Central
Google Scholar
45. Victorino DB, Guimaraes-Marques M, Nejm M, Scorza FA, Scorza CA. COVID-19 and Parkinson’s disease: are we dealing with short-term impacts or something worse? J Parkinsons Dis. 2020;10:899–902.
Article
CAS
PubMed
PubMed Central
Google Scholar
46. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–34.
Article
CAS
PubMed
PubMed Central
Google Scholar
47. DosSantos MF, Devalle S, Aran V, Capra D, Roque NR, Coelho-Aguiar JM, et al. Neuromechanisms of SARS-CoV-2: a review. Front Neuroanat. 2020;14:37.
Article
CAS
PubMed
PubMed Central
Google Scholar
48. Platt MP, Bolding KA, Wayne CR, Chaudhry S, Cutforth T, Franks KM, et al. Th17 lymphocytes drive vascular and neuronal deficits in a mouse model of postinfectious autoimmune encephalitis. Proc Natl Acad Sci USA. 2020;117:6708–16.
Article
ADS
CAS
PubMed
PubMed Central
Google Scholar
49. Sadasivan S, Zanin M, O’Brien K, Schultz-Cherry S, Smeyne RJ. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS ONE. 2015;10:e0124047.
Article
PubMed
PubMed Central
Google Scholar
50. Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007;30:244–50.
Article
CAS
PubMed
Google Scholar
51. Schirinzi T, Martella G, Pisani A. Double hit mouse model of Parkinson’s disease. Oncotarget 2016;7:80109–10.
Article
PubMed
PubMed Central
Google Scholar
52. Rosen B, Kurtishi A, Vazquez-Jimenez GR, Moller SG. The Intersection of Parkinson’s disease, viral infections, and COVID-19. Mol Neurobiol. 2021;58:4477–86.
Article
CAS
PubMed
PubMed Central
Google Scholar
53. Chaudhry ZL, Klenja D, Janjua N, Cami-Kobeci G, Ahmed BY. COVID-19 and Parkinson’s disease: shared inflammatory pathways under oxidative stress. Brain Sci. 2020;10:807.
Article
CAS
PubMed
PubMed Central
Google Scholar
54. Hribar CA, Cobbold PH, Church FC. Potential role of vitamin D in the elderly to resist COVID-19 and to slow progression of Parkinson’s disease. Brain Sci. 2020;10:284.
Article
CAS
PubMed
PubMed Central
Google Scholar
55. Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. 2020;6:eabc5801.
Article
ADS
CAS
PubMed
PubMed Central
Google Scholar
56. Semerdzhiev SA, Fakhree MAA, Segers-Nolten I, Blum C, Claessens M. Interactions between SARS-CoV-2 N-Protein and alpha-Synuclein accelerate amyloid formation. ACS Chem Neurosci. 2022;13:143–50.
Article
CAS
PubMed
Google Scholar
57. Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P. Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci. 2019;42:4–13.
Article
CAS
PubMed
Google Scholar
58. Awogbindin IO, Ishola IO, St-Pierre MK, Carrier M, Savage JC, Di Paolo T, et al. Remodeling microglia to a protective phenotype in Parkinson’s disease? Neurosci Lett. 2020;735:135164.
Article
CAS
PubMed
Google Scholar
59. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69.
Article
CAS
PubMed
Google Scholar
60. McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017;9:14.
Article
PubMed
PubMed Central
Google Scholar
61. Desforges M, Le Coupanec A, Dubeau P, Bourgouin A, Lajoie L, Dube M, et al. Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses. 2019;12:14.
Article
PubMed
PubMed Central
Google Scholar
62. Lang AE, Espay AJ. Disease modification in Parkinson’s disease: current approaches, challenges, and future considerations. Mov Disord. 2018;33:660–77.
Article
PubMed
Google Scholar
63. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18:225–42.
Article
CAS
PubMed
Google Scholar
64. Meinhardt J, Streit S, Dittmayer C, Manitius RV, Radbruch H, Heppner FL. The neurobiology of SARS-CoV-2 infection. Nat Rev Neurosci. 2024;25:30–42.
Article
CAS
PubMed
Google Scholar
65. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92:2105–13.
Article
CAS
PubMed
PubMed Central
Google Scholar
66. Biswas I, Khan GA. Coagulation disorders in COVID-19: role of toll-like receptors. J Inflamm Res. 2020;13:823–28.
Article
CAS
PubMed
PubMed Central
Google Scholar
67. Dosch SF, Mahajan SD, Collins AR. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009;142:19–27.
Article
CAS
PubMed
Google Scholar
68. Xia Y, Zhang G, Kou L, Yin S, Han C, Hu J, et al. Reactive microglia enhance the transmission of exosomal alpha-synuclein via toll-like receptor 2. Brain. 2021;144:2024–37.
Article
PubMed
Google Scholar
69. Sureda A, Alizadeh J, Nabavi SF, Berindan-Neagoe I, Cismaru CA, Jeandet P, et al. Endoplasmic reticulum as a potential therapeutic target for covid-19 infection management? Eur J Pharmacol. 2020;882:173288.
Article
CAS
PubMed
PubMed Central
Google Scholar
70. Chan CP, Siu KL, Chin KT, Yuen KY, Zheng B, Jin DY. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2006;80:9279–87.
Article
CAS
PubMed
PubMed Central
Google Scholar
71. Versteeg GA, van de Nes PS, Bredenbeek PJ, Spaan WJ. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J Virol. 2007;81:10981–90.
Article
CAS
PubMed
PubMed Central
Google Scholar
72. Liao Y, Fung TS, Huang M, Fang SG, Zhong Y, Liu DX. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J Virol. 2013;87:8124–34.
Article
CAS
PubMed
PubMed Central
Google Scholar
73. Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid Redox Signal. 2010;12:839–49.
Article
CAS
PubMed
PubMed Central
Google Scholar
74. Denaro CA, Haloush YI, Hsiao SY, Orgera JJ, Osorio T, Riggs LM, et al. COVID-19 and neurodegeneration: the mitochondrial connection. Aging Cell. 2022;21:e13727.
Article
CAS
PubMed
PubMed Central
Google Scholar
75. Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J. Neurochem. 2009;109:656–69.
Article
CAS
PubMed
Google Scholar
76. Jacobs W, Lammens M, Kerckhofs A, Voets E, Van San E, Van Coillie S, et al. Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail. 2020;7:3772–81.
Article
PubMed
PubMed Central
Google Scholar
77. Wang Y, Huang J, Sun Y, Stubbs D, He J, Li W, et al. SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem Toxicol. 2021;153:112286.
Article
CAS
PubMed
PubMed Central
Google Scholar
78. Kumar A. Experience of video consultation during the COVID-19 pandemic in elderly population for Parkinson’s disease and movement disorders. Postgrad Med J. 2021;97:117–18.
Article
CAS
PubMed
Google Scholar
79. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Article
CAS
PubMed
PubMed Central
Google Scholar
80. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Article
CAS
PubMed
Google Scholar
81. Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022;289:7038–50.
Article
CAS
PubMed
Google Scholar
82. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Article
CAS
PubMed
Google Scholar
83. Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell 2020;181:1188–88 e1.
Article
CAS
PubMed
PubMed Central
Google Scholar
84.Dong-Chen X, Yong C, Yang X, Chen-Yu S, Li-Hua P. Signaling pathways in Parkinson’s disease: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2023;8:73.
Article
PubMed
PubMed Central
Google Scholar
85. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–78.
Article
PubMed
Google Scholar
86. Ito K, Eguchi Y, Imagawa Y, Akai S, Mochizuki H, Tsujimoto Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017;3:17013.
Article
PubMed
PubMed Central
Google Scholar
87. Kabiraj P, Valenzuela CA, Marin JE, Ramirez DA, Mendez L, Hwang MS, et al. The neuroprotective role of ferrostatin-1 under rotenone-induced oxidative stress in dopaminergic neuroblastoma cells. Protein J. 2015;34:349–58.
Article
CAS
PubMed
Google Scholar
88. Devisscher L, Van Coillie S, Hofmans S, Van Rompaey D, Goossens K, Meul E, et al. Discovery of novel, drug-like ferroptosis inhibitors with in vivo efficacy. J Med Chem. 2018;61:10126–40.
Article
CAS
PubMed
Google Scholar
89. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111:16836–41.
Article
ADS
CAS
PubMed
PubMed Central
Google Scholar
90. Jia F, Li H, Jiao Q, Li C, Fu L, Cui C, et al. Deubiquitylase OTUD3 prevents Parkinson’s disease through stabilizing iron regulatory protein 2. Cell Death Dis. 2022;13:418.
Article
CAS
PubMed
PubMed Central
Google Scholar
91. Jia F, Song N, Wang W, Du X, Chi Y, Jiang H. High dietary iron supplement induces the nigrostriatal dopaminergic neurons lesion in transgenic mice expressing mutant A53T human alpha-synuclein. Front Aging Neurosci. 2018;10:97.
Article
PubMed
PubMed Central
Google Scholar
92. Edeas M, Saleh J, Peyssonnaux C. Iron: innocent bystander or vicious culprit in COVID-19 pathogenesis? Int J Infect Dis. 2020;97:303–05.
Article
CAS
PubMed
PubMed Central
Google Scholar
93. Bellmann-Weiler R, Lanser L, Barket R, Rangger L, Schapfl A, Schaber M, et al. Prevalence and predictive value of anemia and dysregulated iron homeostasis in patients with COVID-19 infection. J Clin Med. 2020;9:2429.
Article
CAS
PubMed
PubMed Central
Google Scholar
94. Frazer DM, Anderson GJ. The regulation of iron transport. Biofactors. 2014;40:206–14.
Article
CAS
PubMed
Google Scholar
95. Jia F, Liu H, Kang S. NCOA4-mediated ferritinophagy: a vicious culprit in COVID-19 pathogenesis? Front Mol Biosci. 2021;8:761793.
Article
CAS
PubMed
PubMed Central
Google Scholar
96. Jia FJ, Han J. Liver injury in COVID-19: holds ferritinophagy-mediated ferroptosis accountable. World J Clin Cases. 2022;10:13148–56.
Article
PubMed
PubMed Central
Google Scholar
97. Dufrusine B, Valentinuzzi S, Bibbo S, Damiani V, Lanuti P, Pieragostino D, et al. Iron dyshomeostasis in COVID-19: biomarkers reveal a functional link to 5-lipoxygenase activation. Int J Mol Sci. 2022;24:15.
Article
PubMed
PubMed Central
Google Scholar
98. Ayton S, Lei P, Duce JA, Wong BX, Sedjahtera A, Adlard PA, et al. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann Neurol. 2013;73:554–9.
Article
CAS
PubMed
Google Scholar
99. Wang B, Wang XP. Does ceruloplasmin defend against neurodegenerative diseases? Curr Neuropharmacol. 2019;17:539–49.
Article
CAS
PubMed
PubMed Central
Google Scholar
100. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1alpha/glycolysis-dependent axis. Cell Metab. 2020;32:437–446.e5.
Article
CAS
PubMed
PubMed Central
Google Scholar
101. Cuadrado A, Pajares M, Benito C, Jimenez-Villegas J, Escoll M, Fernandez-Gines R, et al. Can activation of NRF2 be a strategy against COVID-19? Trends Pharmacol Sci. 2020;41:598–610.
Article
CAS
PubMed
PubMed Central
Google Scholar
102. Li X, Zhang Z, Wang Z, Gutierrez-Castrellon P, Shi H. Cell deaths: involvement in the pathogenesis and intervention therapy of COVID-19. Signal Transduct Target Ther. 2022;7:186.
Article
CAS
PubMed
PubMed Central
Google Scholar
103. Mansour HM, Mohamed AF, El-Khatib AS, Khattab MM. Kinases control of regulated cell death revealing druggable targets for Parkinson’s disease. Ageing Res Rev. 2023;85:101841.
Article
CAS
PubMed
Google Scholar
104. Pang Q, Zheng L, Ren Z, Xu H, Guo H, Shan W, et al. Mechanism of ferroptosis and its relationships with other types of programmed cell death: insights for potential therapeutic benefits in traumatic brain injury. Oxid Med Cell Longev. 2022;2022:1274550.
Article
PubMed
PubMed Central
Google Scholar
105. Li G, Wu R, Tong R, Bo B, Zhao Y, Gillen KM, et al. Quantitative measurement of metal accumulation in brain of patients with wilson’s disease. Mov Disord. 2020;35:1787–95.
Article
CAS
PubMed
Google Scholar
106. Ding XS, Gao L, Han Z, Eleuteri S, Shi W, Shen Y, et al. Ferroptosis in Parkinson’s disease: Molecular mechanisms and therapeutic potential. Ageing Res Rev. 2023;91:102077.
Article
CAS
PubMed
Google Scholar
107. Almutary AM, Althunayyan S, Bagalb AS, Mady AF, Alenazi L, Mumtaz SA, et al. Deferoxamine in the management of COVID-19 adult patients admitted to ICU: a prospective observational cohort study. Ann Med Surg. 2023;85:1468–74.
Article
Google Scholar